The fact that our genome encompasses approximately 20,000 genes indicates that the huge variety of existing proteins in the human body cannot be explained by our species’ gene repertory alone. This means that if we want to understand the work of the many players acting in a given dynamic biological system, we should look beyond mere DNA sequencing.
The term transcriptome refers to the entire collection of sequences that are transcribed from DNA into RNA. For protein coding genes, transcriptome analysis provides information on how often a gene is transcribed, how the nascent transcript is processed into mature mRNA, and how stable the messenger (mRNA) is in a cell. The transcriptome is unique to cells, tissues, conditions, and physiological states. The transcriptome of diseased cells has features not found in the transcriptome of their healthy counterparts.
Thus, by analyzing the transcriptome of affected cells or tissues it is possible to identify differences that reveal the many paths leading to disease, offering new opportunities for diagnosing diseases and monitoring their progression.
RNA Analysis by Hybridization-Based Methods
Northern blot was the first laboratory method created to identify specific RNA molecules within a mixture of RNA. Northern blot involves denaturing RNA and separating it by gel electrophoresis, transferring RNA onto a blotting membrane, and hybridizing it with a nucleic acid probe labeled with either a radioactive atom or a fluorescent dye. Although laborious and time-consuming, Northern blot remains a reliable method to study any type of RNA.
Reverse transcription-polymerase chain reaction (RT-PCR) came on the scene after PCR. RT-PCR converts RNA into complementary DNA (cDNA), which then serves as a template for PCR. RT-PCR provides results much faster than Northern blot but quantification can be difficult. Real-time quantitative reverse transcription PCR (qRT-PCR) overcame this limitation, by enabling reliable detection and measurement of products generated during each cycle of the PCR process. Compared to Northern blot, qRT-PCR is easier to perform and requires less time because it’s possible to run the PCR simultaneously at different melting temperatures using a gradient thermocycler. More recently, PCR combined with microfluidic devices has enabled parallel quantification of multiple distinct RNAs.
Despite these advances, the first molecular biology tool capable of quantitating hundreds or thousands of RNAs from a given cell or tissue sample simultaneously was microarray technology. A microarray has thousands of oligonucleotides of known sequences arrayed on a chip, and quantitation relies upon hybridization of sample RNA that has been reverse-transcribed and labeled.
Although microarray technology flourished to become the main platform for high-throughput analysis of RNA in biological systems, it shares a major limitation with all other hybridization-based methods in that it requires previous knowledge of the RNA molecules to be analyzed, thus limiting the potential for discovery.
Additional drawbacks of hybridization-based approaches include high background levels resulting from cross-hybridization and a limited dynamic range of detection due to both background and saturation of signals. For these reasons, transcriptomic analysis based on microarrays typically failed to provide relevant information that could be transferred directly into clinical applications.
The Advent of RNA-Seq
In contrast to hybridization methods, sequence-based approaches directly determine the cDNA sequence. Initially, Sanger sequencing of cDNA was used, but following the development of next-generation sequencing (NGS) and deep-sequencing technologies, RNA sequencing (RNA-Seq) emerged and revolutionized the way entire transcriptomes would be analyzed.
NGS differs from conventional capillary-based (Sanger) sequencing in being able to process millions of sequence reads in parallel. Deep sequencing is an NGS approach that sequences the same cDNA region hundreds or even thousands of times.
The RNA-Seq workflow (Fig. 1) starts with selecting a method for RNA purification best adapted to the biological sample being analyzed. After assessing the quantity and quality of the purified RNA, the next step consists of converting the population of RNA molecules into a library of cDNA fragments. This involves a reverse transcriptase-mediated first strand synthesis followed by a DNA polymerase-mediated second strand synthesis.
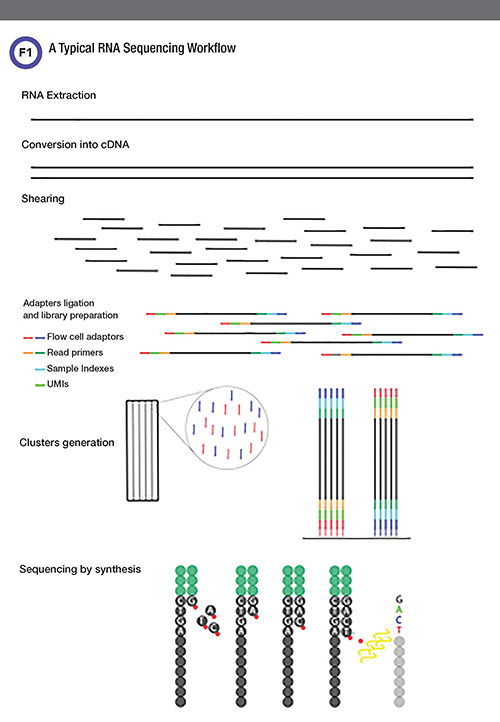
In a typical RNA-Seq experiment, RNAs are converted into a library of cDNA fragments of an appropriate size for sequencing through either RNA fragmentation or complementary DNA (cDNA) fragmentation. Sequencing adaptors are added to each fragment. Each fragment is further tagged with a unique molecular identif¬ier (UMI) sequence chosen from approximately 10,000 combinations, so that two identical molecules become distinguishable; this provides a digital measurement of absolute numbers of each RNA species, irrespective of PCR amplification biases. Index sequences are also introduced, enabling sequencing up to 96 different samples on a single run. In the Illumina sequencing platform, oligonucleotides complementary to the adaptor sequences are immobilized on the surface of a flow cell. Attached cDNA fragments anneal to a nearby read primer for polymerase chain reaction (PCR) amplification. Repeated cycles form colony-like clusters, each containing approximately 1,000 copies. The sequencing reaction (sequencing-by-synthesis) is then carried out with fluorescently labeled modified nucleotides that act as reversible terminators. Thus, only a single fluorescent nucleotide can be added by a polymerase to each growing DNA copy. Images are recorded to identify the fluorescent base incorporated in each cluster. Next, the fluorophores are cleaved off and the terminators are removed, allowing another round of nucleotide incorporation.
Depending on the NGS sequencing platform, specific adapter sequences are attached to one or both ends of each cDNA fragment. Short (typically 50-150bp long) sequence reads are then obtained from either one end (single-end sequencing) or both ends (pair-end sequencing) of each cDNA. Finally, bioinformatics methods are used to align the individual reads to the reference genome or transcriptome.
RNA-Seq provides more precise qualitative and quantitative information than previous technologies and has already reshaped our view of several organisms’ transcriptomes. RNA-Seq has revealed many novel transcribed regions in every genome analyzed, from yeast to human, as well as many novel human mRNA isoforms resulting from alternative splicing, alternative polyadenylation, and/or alternative promoter use. Moreover, profiling the transcriptome of thousands of human cancer samples has unraveled specific gene fusions and gene expression signatures with demonstrated prognostic and predictive value.
RNA-Seq: Maturing Method for Diagnosis
The current rate of molecular diagnosis of Mendelian diseases is low and between 25% and 50%. The vast majority of known genetic alterations associated with Mendelian disorders have been identified by sequencing the protein-coding regions of genes (exome sequencing). However, evidence is mounting that hereditary diseases can also be caused by mutations located within non-coding regions of the genome, such as introns, transcriptional regulatory sequences of protein-coding genes, and non-coding regulatory RNA genes (1).
Mutations leading to down-regulation of gene expression can be better identified by combining DNA and RNA sequencing (2). The definitive functional identification of mutations that interfere with splicing also requires RNA analysis. Indeed, the correct removal of introns from the nascent precursor mRNAs is a tightly controlled process that depends upon combinatorial cross-talk between the splice site sequences and regulatory sequences located within exons and introns. Thus, not only mutations located deep within introns (that are not detected by exome sequencing), but also exonic mutations may alter splicing. Identification of both intronic and exonic mutations affecting splicing will increase the overall mutation-detection rates, which ultimately will facilitate more patients carrying disease-causing mutations to receive the right diagnosis.
However, a major downside of using RNA-Seq for human studies is that gene expression is cell-type specific. Thus, it may be necessary to extract RNA from the tissue or organ that is specifically affected in a particular disease, and many tissues in the human body are not easy to access.
Despite this drawback, RNA-Seq is starting to offer new hopes for many patients and families carrying a hereditary disease for which DNA sequencing failed to provide a diagnosis (3). For instance, a recent study (4) used RNA-Seq to analyze RNA extracted from muscles obtained from biopsy samples of 63 patients with a range of muscle disorders. While some patients already had a defined diagnosis and were included in the study to validate the findings obtained with RNA-Seq, others had not yet received a diagnosis. RNA-Seq was able to correctly determine the molecular diagnosis for 66% of patients whose samples already had undergone DNA sequencing and for which strong gene candidates were indicated.
In contrast, RNA-Seq identified aberrant splicing isoforms and provided a diagnosis in 21% of cases lacking candidate mutations. Overall, the researchers made 17 new diagnoses and associated the splicing abnormalities with mutations located either at splice sites or within introns that caused exon extension (when the exon extends beyond its normal limits), intronic splice gain (gain of intronic sequence), exon skipping (when an exon is missing in the transcript), and other splice disruptions. Of note, without the RNA-Seq analysis these genetic variants would be reported as variants of unknown significance (VUS).
Another study (5) analyzed fibroblasts from 105 patients with suspected mitochondrial disease. Almost half of the patients (48) submitted samples for whole-exome sequencing but no molecular diagnosis could be established. RNA-Seq analysis revealed aberrant splicing isoforms by comparing the pattern of each patient against the others.
One patient showed a splice defect that resulted in a truncated CLPP, a mitochondrial ATP-dependent endopeptidase, and Western blot analysis confirmed the loss of the full-length CLPP. The variant, detected as a homozygous change in the very last nucleotide of exon 5 of the CLPP gene, had been previously reported as VUS.
Based on the results obtained by RNA-Seq, this variant was reclassified as disease-causing. Knowing that CLPP was the implicated gene allowed clinicians to associate the patient’s manifestations with Perrault syndrome (OMIM #601119), which is caused by a deficiency of the CLPP protein.
In another patient, whole-exome sequencing revealed a mono-allelic expressed VUS in the ALDH18A1 gene; this variant was a compound heterozygous with a nonsense variant in the same gene. RNA-Seq showed very low levels of ALDH18A1 mRNA. Quantitative proteomics confirmed that the levels of the ALDH18A1 protein were almost zero, and functional studies supported the reclassification of the two detected variants as disease-causing.
Hamanaka et al. recently proposed a workflow strategy to combine exome sequencing and RNA-Seq findings to solve undiagnosed cases of patients with nemaline myopathy (NM)(6). Skin samples from six patients with incomplete molecular diagnosis were submitted to the workflow. In these cases, exome sequencing had revealed only one of the pathogenic variants between the two required to cause NM, but RNA-Seq solved four cases by identifying the second missing pathogenic allele. In all four cases the second hit involved abnormal splicing events.
Conclusion
RNA-Seq is solving several undiagnosed cases for which DNA sequencing alone was inconclusive. However, a major downside of using RNA-Seq to diagnose hereditary diseases is that in many cases RNA needs to be extracted from the affected tissue or organ because expression of the mutated gene is tissue-specific.
Nevertheless, evidence is mounting that combined DNA and RNA analysis may greatly increase the success rate of molecular diagnosis of hereditary diseases, offering new opportunities for discovery and new hopes for families affected by hereditary genetic disorders.
Catarina Silveira, MSc,is a senior molecular genetic laboratory technologist at GenoMed Diagnostics, in Lisbon, and aPhD candidate at the University of Lisbon in Portugal. +Email: [email protected] Lobo Antunes, University of Lisbon Medical School, Portugal. +Email: [email protected]
Maria Carmo-Fonseca, MD, PhD, is a principal investigator at Instituto de Medicina Molecular João Lobo Antunes, University of Lisbon Medical School, Portugal. +Email: [email protected]
References
- Vaz-Drago R, Custódio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet 2017;136:1093.
- Byron SA, Keuren-jensen KR Van, Engelthaler DM, et al. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat Rev Genet 2016;17:257-71.
- Chakravorty S, Hegde M. Clinical utility of transcriptome sequencing: Toward a better diagnosis for Mendelian disorders. Clin Chem 2018;64:882-4.
- Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:eaal5209.
- Kremer LS, Wortmann SB, Prokisch H. Transcriptomics: Molecular diagnosis of inborn errors of metabolism via RNA-sequencing. J Inherit Metab Dis 2018;41:525-32.
- Hamanaka K, Miyatake S, Koshimizu E, et al. RNA sequencing solved the most common but unrecognized NEB pathogenic variant in Japanese nemaline myopathy. Genet Med 2019; 21:1629–38.