Hemoglobinopathies and thalassemias involve problems with hemoglobin, the critical protein in red blood cells (RBC) responsible for transporting oxygen throughout the body. Abnormal hemoglobin, present in sickle cell diseases, or too little hemoglobin, such as may occur in the thalassemias, can cause organ damage and shorten patients’ lifespans. Hemoglobin consists of 4 globin polypeptide chains. Adult hemoglobin A consists of 2 alpha chains and 2 beta chains (α2β2). The α chain comprises 141 amino acids; the β chain 146.
There are many disorders of hemoglobin assembly and function, including at least three broad types. First, inherited disorders of heme biosynthesis include the porphyrias and X-linked sideroblastic anemia. Second are states where the heme can no longer bind to oxygen, such as when the ferrous heme iron is oxidized to the ferric form (inherited or acquired methemoglobinemia) or when heme complexes with carbon monoxide (carboxyhemoglobin). Collectively, such states are called dyshemoglobins. Third are the inherited abnormalities of the globin polypeptide biosynthesis, structure, and function.
This review will focus on the latter group. Within this subset, there are two overlapping groups: the hemoglobinopathies and the thalassemias. Hemoglobinopathies typically have at least one amino acid substitution leading to synthesis of a variant globin chain.
Thalassemias, on the other hand, involve perturbation of the rate of globin chain synthesis (1). This leads to a relative excess synthesis of the normal globin chain. Clinically, a thalassemia trait is termed thalassemia minor. More severe forms are known as intermedia or major depending on whether or not patients are transfusion-dependent.
Analysis of Hemoglobin
Electrophoresis in an alkaline environment, most commonly on an agarose gel, still remains an important means of hemoglobin variant identification (1–3). Following electrophoresis, the hemoglobin bands can be visualized with a protein stain (Figure 1). Hb A (α2β2), which makes up more than 95% of hemoglobin in normal adults, migrates most rapidly toward the anode (+). The only other hemoglobin band that is visible on alkaline gel electrophoresis in normal adults and children (not neonates) is Hb A2 (α2δ2) which is present at low concentrations, < 3.5% of total hemoglobin. Blood from healthy newborns typically shows 20–30% Hb A, 70–80% hemoglobin F (α2γ2), and no Hb A2.
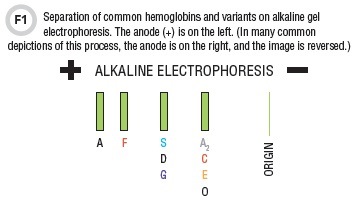
On alkaline gels the order of migration for the normal hemoglobins as well as the most common hemoglobinopathies is A, F, S, and A2/C (Figure 1) (1–3). Alkaline gel electrophoresis does not produce an accurate or precise Hb A2 quantitation. To quantitate Hb A2, clinical laboratories have to use cation exchange high-performance liquid chromatography (CE-HPLC) or capillary zone electrophoresis (CZE).
The migration position on alkaline gels is not completely unique. Hemoglobins G and D, if present, will migrate with hemoglobin S, while hemoglobin E migrates in the same position as C and A2. For this reason, there is another diagnostic modality—acid gel electrophoresis (1–3). On acid gels, hemoglobins A and A2 migrate together with D, G, and E, but C, S, and F occupy relatively unique positions (Figure 2).

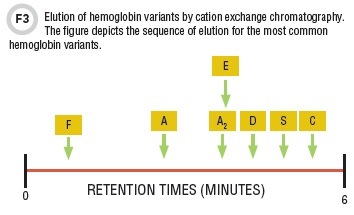
Another means of analysis is CE-HPLC (Figure 3). The hemoglobins are eluted by an ionic gradient and detected spectrophotometrically at 415 nm. Importantly, Hb A2 can be accurately quantitated. Typically, whole blood specimens with no pre-preparation can be added to the automated sampler.
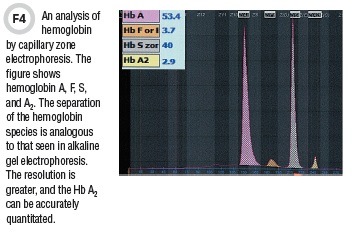
CZE has been in use for some time in the field of serum protein separation, and in the last decade this technique has been adapted for hemoglobin separation. Separation occurs by electrophoresis in a long, but very narrow-bore silica capillary tube. The capillary is filled with sample and buffer and extends between two buffer reservoirs. Separation is effected by application of a very high voltage. The hemoglobin species are detected optically, much the same way as HPLC. CZE of hemoglobins produces a profile similar to the alkaline separation in traditional gels, but at a much higher resolution (Figure 4). Like CE-HPLC, this technique produces a precise Hb A2 quantitation.
Hemoglobin Variants: S, C, and E
Individuals who are heterozygous for hemoglobin S (Hb AS or sickle trait) are asymptomatic and have a normal peripheral smear. About 8% of African-Americans carry the sickle trait. In contrast, Hb S homozygotes suffer from sickle cell disease or sickle cell anemia. This condition results from a glutamic acid-to-valine switch in the 6th amino acid of the beta chain (b6 Glu-→Val). Hemoglobins containing the βS polypeptide chain, when in the deoxygenated form, are far less soluble than normal adult Hb A. The insoluble deoxyhemoglobin S polymerizes and makes the RBC very rigid. This sickled RBC is unable to traverse the microcirculation, resulting in obstruction of small blood vessels.
S homozygotes typically produce Hb F at relative percentages that range from 0% up to 20% in untreated individuals. Individuals who are AS heterozygotes (sickle trait) show both Hb A and Hb S in the ratio of approximately 60:40, with normal concentrations of Hb A2. Hemoglobins D, GPhiladelphia, and Korle-Bu migrate in the same position as Hb S.
In new cases of sickle trait or sickle disease, either acid gel electrophoresis and/or HPLC should be performed to positively identify the variant.
Hemoglobin C
Hemoglobin C heterozygosity, or C trait, is a silent condition that is seen in about 2–3% of African-Americans and is associated with a low-normal mean corpuscular volume (MCV). On peripheral smear, there may be normocytosis or mild microcytosis with target cells. In the homozygous state (βC/βC), there is a mild hemolytic anemia. The microcytosis in homozygotes may be striking and there are abundant target cells. The mutation that produces hemoglobin C occurs at the same position (β6) as the S mutation, but in this case is β6 Glu- →Lys.
Hemoglobin SC Disease
About 0.13% of African-Americans are Hb SC compound heterozygotes. Hb SC results in a sickling disorder similar to Hb S homozygous state. These individuals may be mislabeled as having homozygous S disease. Hemoglobin electrophoresis shows both Hb S and Hb C with no hemoglobin A.
Hemoglobin E
Hemoglobin E (β26 Glu- →Lys) is common in Southeast Asia. The presence of Hb E may be associated with a thalassemic phenotype since the mutated beta chain is synthesized more slowly than normal. Homozygosity, however, is very mild, and most patients are asymptomatic. Homozygotes have a mild anemia with a reduced MCV and an elevated RBC count. Hb E heterozygotes tend to show a reduced MCV but usually with no anemia or they have a minimal reduction in hemoglobin.
Thalassemias
Thalassemia traits are typically associated with a mild or borderline anemia as well as uniform RBC microcytosis and an elevated or high-normal RBC count. More severe thalassemias may show a moderate-to-severe anemia with a marked microcytosis.
Beta thalassemias (1)
Beta thalassemias are usually due to point mutations in or near the beta globin gene on chromosome 11. The mutations may affect the gene promoter sequence, the 5’ untranslated sequence (UTR) of the coding region, the initiation codon, or the mutations may involve RNA processing, leading to inefficient or absent splicing. β+ mutations result in a reduced (but not absent) rate of synthesis of beta polypeptide chains and range from severe to mild, while in β0 mutations, there is no detectable beta globin production from the mutated allele.
Beta thalassemia traits refer to either the β/β+ or the β/β0 genotype. These are clinically mild conditions that result in microcytosis, mild anemia, and an elevated RBC count. The beta thalassemia homozygote or compound heterozygote state results in a more severe phenotype. The β0/β0, β+/β+, or β+/β0 state in which the beta plus (β+) mutation is severe, will produce a transfusion-dependent thalassemia major, while the β+/β+ genotype involving the milder beta mutations may produce a thalassemia intermedia.
The diagnostic hallmark of the beta thalassemia trait on hemoglobin analysis is an elevated relative percentage of Hb A2. In this condition, the Hb A2 is typically 4–8% with a mean of about 5–6%. However, elevation of Hb A2 is not universal in all beta thalassemia traits. A notable exception is beta thalassemia with coexistent iron deficiency, in which iron deficiency may suppress or mask the Hb A2 elevation, so the percentage Hb A2 is best judged following iron repletion. There may also be a slight elevation of Hb F in beta thalassemia traits. This elevation is not usually more than 5–7% of the total hemoglobin.
Beta thalassemia homozygotes or compound heterozygotes have a more marked anemia in which Hb F may be the dominant hemoglobin. The relative percentage of adult Hb A varies from 0% in the β0/β0 state to over 30% in milder β+/β+ variants.
Alpha Thalassemias (1, 4)
Normal individuals carry a total of four α globin genes, and α thalassemias are most commonly a consequence of gene deletions. Deletions may affect the two α genes on the same chromosome (i.e., in cis) or α genes on the homologous chromosome (trans) (Figure 5).
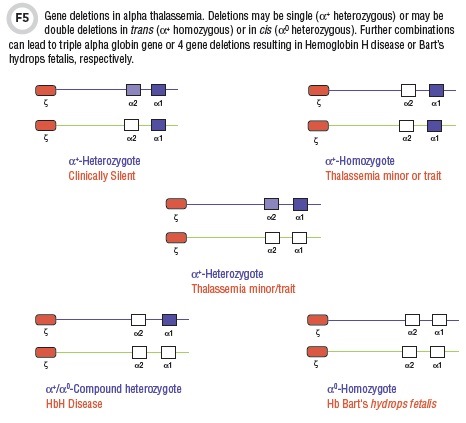
The two major deletional genotypes are α+ and α0 (4). The α+ genotype refers either to the loss of a single alpha globin gene (α+ heterozygote) or two alpha genes in trans (α+ homozygote) and is found in individuals of African ancestry, as well as those of Mediterranean, Thai, or Indonesian ethnic backgrounds. The α0 genotype exists when 2 α genes are deleted in cis (Figure 5) (4).
The -α/αα condition is completely silent with minimal or no changes on peripheral smear. -α/-α or --/αα individuals—both have two gene deletions—have a thalassemic phenotype with RBC microcytosis, a mild anemia, and a disproportionately elevated RBC count i.e., thalassemia trait. In such conditions, the hemoglobin analysis by all modalities is entirely normal. The Hb A2 fraction is not increased and there is no detectable Hb F.
A more severe phenotype results from the α+/α0 compound heterozygous state, resulting in a three gene deletion (--/-α) genotype (Figure 5). Affected patients, in general, have a moderate-to-severe anemia with a prominent microcytosis. Hemoglobin electrophoresis shows a very rapid migrating band (Hb H) on alkaline gel electrophoresis or CZE and an early-eluting peak on HPLC. Hb H consists of four β chains.
The most severe phenotype is
the four gene deletion alpha thalassemia which occurs in α0 homozygotes (--/--). This is incompatible with life, and results either in stillbirth or rapid demise after birth. The syndrome is called Hb Bart’s hydrops fetalis and is associated with severe intrauterine anemia, cardiac failure, and multiple effusions. Analysis of cord blood is very characteristic.
The majority of the hemoglobin (80–100%) is Hb Bart’s, which is made up of four g chains.
Although the preceding discussion delineates 2 genotypes, α+ and α0, these are not homogeneous groups. There are seven major deletional genotypes referred to respectively as -α3.7, -α4.2, -(α)20.5, --SEA, --MED, --FIL, and --THAI. -α3.7 and -α4.2 produce the single gene α+ type, while -(α)20.5, --SEA, --MED, --FIL, and --THAI produce the two gene α0 genotype.

References
- Harris NS, Winter WE. Hemoglobinopathies: Biochemical disorders of hemoglobin. In: Clarke, W. Contemporary practice in clinical chemistry. 2nd Ed. Washington, D.C.: AACC Press 2011;249–63.
- Bain BJ. Hemoglobinopathy diagnosis. Oxford, London, Edinburgh (U.K.), Malden, Massachusetts (U.S.): Blackwell Science Ltd.; 2001.
- Hoyer JD, Kroft SH, eds. Color atlas of hemoglobin disorders: A compendium based on proficiency testing. Northfield, Illinois: College of American Pathologists (CAP); 2003.
- Piel FB, Weatherall DJ. The α-Thalassemias. N Engl J Med 2014;371:1908–16.
Neil Harris, MD is an assistant professor of coagulation and unit director of coagulation and chemistry at the University of Florida in Gainesville.
+Email: [email protected]
William Winter, MD is a professor of pathology and unit director of endocrinology at the University of Florida in Gainesville.
+Email: [email protected]