
Access to routine HIV testing and linkage to care still pose significant barriers to HIV treatment and prevention (3). Globally, there are approximately 38 million people living with HIV, with an estimated 1.2 million in the United States (1). In the most recent surveillance report, the Centers for Disease Control and Prevention (CDC) estimated that approximately 13% of cases (156,000) remain undiagnosed, and 40% of new infections occur because of unknown HIV status (2).
To tackle this public health priority, the CDC launched the Ending the HIV Epidemic initiative, aiming to eliminate the HIV epidemic by the end of 2030. A core tenet is to prevent forward viral transmission by leveraging current and emerging testing technologies (4).
In May 2022, the United States Food and Drug Administration (FDA) took a step forward along this path and announced that it would reclassify HIV diagnostic tests from Class III to Class II medical devices (13). This is the first instance of medical device reclassification in the HIV testing landscape. Renewed regulatory classification of HIV diagnostic assays may have implications for time, cost, and new analytical developments, and may lead to increased testing availability.
Current tests include antibody (Ab), antibody/antigen (Ab/Ag), and RNA based testing. The latest CDC testing algorithm begins with an FDA-approved Ag/Ab immunoassay. Laboratories confirm positive results through a secondary method, starting with a supplemental antibody immunoassay that differentiates HIV-1 from HIV-2 antibodies (8).
Providing increased access to simple and routine testing empowers all individuals to know their status while destigmatizing testing. Identifying infections early and connecting people to treatment are proven to reduce HIV transmission, as shown in clinical trials and large community-based interventions (5–7). Developing and deploying HIV testing is instrumental in reducing global HIV incidence.
The Evolution of HIV Testing
Since 1985, when the first HIV screening test was designed, there have been several major methodologic improvements. The generations of HIV immunoassays are summarized in Table 1. A significant recent advancement was adding p24 antigen testing to immunoassay screens, which detects HIV-1 infection earlier than HIV antibodies alone. The p24 antigen, an HIV-1 capsid protein, appears within approximately 2 weeks of HIV infection and is a marker of recent HIV infection.
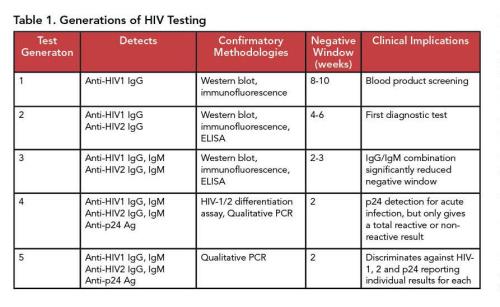
However, as antibodies to the viral capsid protein are produced, p24 antigen levels become undetectable. This makes fourth and fifth generation assays particularly useful in populations with a higher proportion of recent infections.
While CDC recommends a combined Ab/Ag assay, third generation assays that detect antibodies against HIV-1 and HIV-2 remain the main technology used in point-of-care testing (POCT). Several feasibility studies have demonstrated the suitability and clinical utility of POCT and self-testing in community-based HIV mitigation strategies (10–12). However, OraQuick, the only FDA-approved home test—as well as the majority of POCT—is based on third-generation technology. This underscores the clinical need for simple, deployable technology capable of detecting recent infections (9).
FDA’s action to reclassify HIV tests has the potential to speed newer technology to the point of care.
A New Phase Of HIV Test Development
Before the agency’s 2022 decision, FDA classified all HIV diagnostic tests under its highest risk classification. The European Union still classifies medical devices for HIV testing as class D testing, which is the highest regulatory risk category (14).
Traditionally, diagnostic assays are classified as either Class III or Class II devices. Class III, as the highest risk classification, encompasses devices that are life-sustaining or critical to health. At least one clinical trial is required to demonstrate utility and efficacy. Test manufacturers package these in a premarket approval (PMA) submission to the FDA.
PMAs require prospective enrollment in clinical trials, which is not only time- and cost-intensive but also is influenced by the target population for study inclusion. Results from the trial, including device performance and utility, are supposed to be readily translatable to a general population. This is a major strength of class III regulatory policy.
With HIV testing specifically, it will be important for new assays to be designed that can identify specific HIV subtypes and/or recent versus long-term infections. With the global decline in HIV incidence, recruitment of serodiverse individuals has become increasingly difficult, requiring larger cohorts and more time to assess the range of clinically relevant test results.
Class II devices are categorized as intermediate risk and do not require prospective clinical trials to demonstrate efficacy. Class II devices undergo a 510(k) review in which the device must demonstrate equivalence to another device on the market.
Regulatory requirements for Class II devices are largely focused on analytical performance and do not assess clinical utility. Further, Class II devices may be regulated with the addition of special controls that are specific to device type. This is the approach FDA is taking with HIV testing.
The new regulatory guidance for HIV device special controls includes strict requirements for labeling, documentation, and data submission indicating a change in performance characteristics, cross-method validation, and result interpretation (13).
Class II devices must meet the same analytical criteria as class III devices. Manufacturers often evaluate the performance of class II devices using specimens from a biorepository rather than prospective clinical trials. Analysis of banked specimens allows the process to include both acute and more established HIV infections, as well as analysis in specific populations, such as those with other comorbidities or sexually transmitted infections. However, the FDA reclassification does not provide prescriptive details about specimen number or sample characteristics required during validation, other than what is necessary to meet performance criteria.
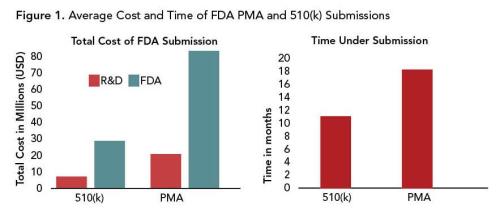
In a survey of more than 200 medical device companies, the average total cost of bringing a Class III device to market through the PMA pipeline, including research and development, was approximately $91 million. Cost estimates from the FDA submission process alone range from $54–$74 million (15, 16). In contrast, the costs for a 510(k) submission average $25 million, a reduction of 50–75% (Figure 1A).
This same survey also reported that the PMA submission process lasted 18 months, as opposed to the 510(k) process, which averaged 11 months (16) (Figure 1B). Time and costs associated with regulatory submission are frequently cited as the two largest factors that hinder medical device innovation.
FDA approval serves as an important international benchmark of quality. Thus, laborious and administratively burdensome regulatory processes can lead to delayed approval and availability of medical technologies to patients in the U.S. and abroad. On average, a new medical device will become available in the U.S. approximately 2 years after the same device is available in Europe due to decreased regulatory burden (17).
From a cost and time perspective, changes in the FDA classification of HIV testing likely will incentivize medical device development internationally.
Bringing Better HIV Tests to Market
As new devices move through the regulatory process, representative sample analysis is integral to robust and translatable technology. Devices should be developed in relation to the populations they serve and should capture both the diversity of the population—considering factors such as geographic location, age, and background—as well as serodiversity. Serodiversity encompasses samples from HIV seronegative and HIV seropositive individuals, as well as individuals infected with different HIV-1 groups and subtypes.
Assay development with banked specimens can affords manufacturers the opportunity to assess device performance in a more inclusive and equitable fashion; however, the choice to do so remains with the manufacturer. This underscores the importance of strengthening global initiatives and supporting public health surveillance to advance equity in medicine.
The HIV epidemic remains a global public health priority, with the largest impact in historically underrecognized. Revising policy and incentivizing innovation will assist in the fight against HIV, but these efforts will not succeed without recognizing, including, and prioritizing those individuals who are most at risk.
Ultimately, the regulatory reclassification of HIV diagnostic devices opens the door for larger conversations surrounding increased deployment and implementation of medical devices to areas with the highest need, supporting the global mission to reduce, and ultimately eliminate, HIV transmission.
Ashley R. Rackow, PhD, is a postdoctoral fellow in the department of pathology at Johns Hopkins University School of Medicine in Baltimore. +Email: [email protected]
Mark A. Marzinke, PhD, DABCC, FADLM, is a professor of pathology and medicine at the Johns Hopkins University School of Medicine, director of general chemistry in the Johns Hopkins Hospital Core Laboratories, and director of the clinical pharmacology analytical laboratory within the division of clinical pharmacology. He is also the co-principal investigator of the HIV Prevention Trials Network Laboratory Center in Baltimore. +Email: [email protected]
References
- UNAIDS. Global AIDS Update: Seizing the Moment. Geneva; 2020.
- Centers for Disease Control and Prevention. Estimated HIV incidence and prevalence in the United States, 2015–2019. HIV Surveillance Supplemental Report 2021; 26 (No. 1). http://www.cdc.gov/ hiv/library/reports/hiv-surveillance.html (Accessed May 2023).
- Centers for Disease Control and Prevention. HIV/AIDS: Basic Statistics. https://www.cdc.gov/hiv/basics/statistics.html (Accessed May 2023).
- Centers for Disease Control and Prevention. Ending the HIV Epidemic in the U.S. https://www.cdc.gov/endhiv/index.html (Accessed May 2023).
- Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 Infection with Early Antiretroviral Therapy. N Engl J Med 2011; doi: 10.1056/NEJMoa1105243.
- Bavinton BR, Pinto AN, Phanuphak N, et al. Viral suppression and HIV transmission in serodiscordant male couples: an international, prospective, observational, cohort study. Lancet HIV 2018; doi: 10.1016/S2352-3018(18)30132-2
- Rodger AJ, Cambiano V, Bruun T, et al. Sexual activity without condoms and risk of HIV transmission in serodifferent couples when the HIV-positive partner is using suppressive antiretroviral therapy. JAMA 2016; doi: doi:10.1001/jama.2016.5148.
- National Center for HIV/AIDS VH and TPD of HP. Recommended laboratory HIV testing algorithm for serum or plasma specimens. 2018.
- Bouzid D, Zanella MC, Kerneis S, et al. Rapid diagnostic tests for infectious diseases in the emergency department. Clin Microbiol Infect 2021; doi: 10.1016/j.cmi.2020.02.024.
- Devillé W, Tempelman H. Feasibility and robustness of an oral HIV self-test in a rural community in South-Africa: An observational diagnostic study. PLoS One 2019; doi: 10.1371/journal.pone.0215353.
- Sanabria G, Scherr T, Garofalo R, et al. Usability evaluation of the mLab app for improving home HIV testing Behaviors in youth at risk of HIV infection. AIDS Educ Prev 2021; doi: 10.1521/aeap.2021.33.4.312.
- Zelin J, Garrett N, Saunders J, et al. An evaluation of the performance of OraQuick ADVANCE Rapid HIV-1/2 Test in a high-risk population attending genitourinary medicine clinics in East London, UK. Int J STD AIDS 2008; doi: 10.1258/ijsa.2008.008132.
- Food and Drug Administration. Reclassification of Human Immunodeficiency Virus Serological Diagnostic and Supplemental Tests and Human Immunodeficiency Virus Nucleic Acid Diagnostic and Supplemental Tests. Federal Register 2022.
- van Norman GA. Drugs and Devices Comparison of European and U.S. Approval Processes. 2016.
- Sertkaya A, Devries R, Jessup A, Beleche T. Estimated Cost of Developing a Therapeutic Complex Medical Device in the US. JAMA Netw Open 2022; doi:10.1001/jamanetworkopen.2022.31609.
- Makower JMADL. FDA Impact on U.S. Medical Technology Innovation. 2010.
- Sorenson C, Drummond M. Improving medical device regulation: The United States and Europe in perspective. Milbank Q 2014; doi: 10.1111/1468-0009.12043.