Epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor that plays a central role in regulating cell division and death. It belongs to the HER family of receptors, which includes EGFR (HER1/ErbB1), ERBB2 (HER2/neu), ERBB3 (HER3), and ERBB4 (HER4). These receptors are characterized by an extracellular ligand-binding domain, a transmembrane domain, and a cytoplasmic domain containing the tyrosine kinase region followed by a carboxy-terminal tail with tyrosine autophosphorylation sites. Several growth factors, including EGF, transforming growth factor-α (TGF-α), amphiregulin (AR), epiregulin (EREG), heparin-binding EGF (HB-EGF), betacellulin (BTC), and epigen (EPG) bind to the ErbB receptors.
Mutations in the gene encoding EGFR that lead to overexpression of the protein have been associated with a number of different cancers. Furthermore, this upregulation appears to be frequently associated with adverse prognosis. These discoveries have stimulated researchers to identify novel biomarkers that can be used to determine which patients have the mutations so that they can receive therapeutics targeted to their cancer.
Here I present an overview of the EGFR signaling pathway, as well as testing and therapeutic strategies for patients who test positive for various mutations in the signaling pathway.
EGFR Signaling Pathway
Following ligand binding at the extracellular domain, EGFR forms functionally active homo- or hetero-dimers. Dimerization induces the activation of the tyrosine kinase domain, leading to autophosphorylation of the receptor on multiple tyrosine residues. This phosphorylation triggers recruitment of a range of adaptor proteins, such as Src homology 2 domain containing (SHC) protein and growth factor receptor-bound protein 2 (GRB2), followed by activation of a series of intracellular signaling cascades that affect cell proliferation, apoptosis, invasion, metastasis, and angiogenesis.
Downstream effects of EGFR activation include modulation of three major pathways that have been well characterized (Figure 1). Induction of the RAS-RAF-MAPK pathway occurs when phosphorylated EGFR recruits the guanine-nucleotide exchange factor via the GRB2 and SHC adapter proteins, which activates RAS. This step subsequently stimulates RAF and the MAP kinase pathway, ultimately affecting cell proliferation, tumor invasion, and metastasis.
Figure 1
EGFR Signaling Pathway
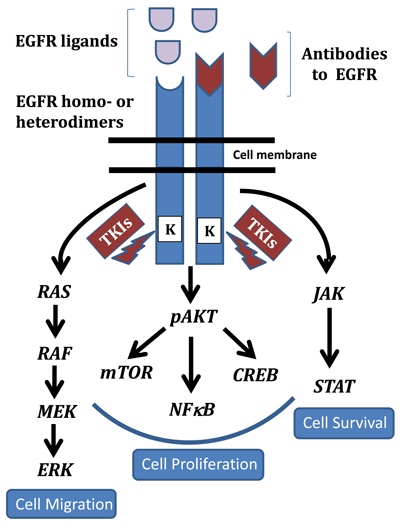 EGFR is activated either as a homo- or heterodimer resulting in regulation of multiple pathways. In particular the RAS/RAF/MAPK, AKT, and JAK/STAT pathways downstream of EGFR play integral roles in cell migration, proliferation, and survival, respectively. Anti-EGFR antibodies are targeted to the external ligand binding domains while the small molecule inhibitors or tyrosine kinase inhibitors (TKIs) target its cytoplasmic kinase domains.
EGFR is activated either as a homo- or heterodimer resulting in regulation of multiple pathways. In particular the RAS/RAF/MAPK, AKT, and JAK/STAT pathways downstream of EGFR play integral roles in cell migration, proliferation, and survival, respectively. Anti-EGFR antibodies are targeted to the external ligand binding domains while the small molecule inhibitors or tyrosine kinase inhibitors (TKIs) target its cytoplasmic kinase domains.
Following activation of the EGFR pathway, the phosphatidylinositol 3-kinase (PI3K/AKT) pathway induces the major cellular survival and anti-apoptosis signals by stimulating nuclear transcription factors such as NFKB. Involvement of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway also is implicated in activating transcription of genes associated with cell survival.
EGFR activation affects other not so commonly known pathways as well. These include phosphorylation of phospholipase C gamma 1(PLCG) and subsequent hydrolysis of phosphatidylinositol 4,5 biphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), which results in activation of protein kinase C (PRKC). Activation of this kinase turns on the MAPK and c-Jun kinase pathways.
Today we know that EGFR signaling plays an essential role in cell proliferation, survival, and migration. Activating mutations of the EGFR gene are found predominantly in females, individuals who have never smoked, and individuals with Asian ethnicity. Though its altered activity has been studied primarily in development and growth of non-small cell lung cancer (NSCLC), many other tumors, including head and neck, ovary, cervix, bladder, esophagus, stomach, brain, endometrium, colon, breast, and liver are also known to exhibit deregulation of EGFR.
Therapeutic Strategies Targeting EGFR
Gene amplification and over-expression of the Erb family of receptors (EGFR and ErbB2) has been observed in breast, lung, and colorectal cancers, while the deregulated activation of intracellular mitogenic signaling has been implicated in many other cancers. These two pathways make protein target-based therapies very promising tools for treatment.
In fact, pharmaceutical companies have developed several potential therapies that use anti-EGFR monoclonal antibodies (mAbs) and small-molecule tyrosine kinase inhibitors (TKIs) that target distinct domains of EGFR. Anti-EGFR monoclonal antibodies, such as cetuximab, panitumumab, and nimotuzumab, bind to the extracellular domain of the EGFR monomer and compete for receptor binding by the endogenous ligands, triggering receptor internalization and blocking ligand-induced receptor activation. Cetuximab, which binds to the L2 domain of EGFR, is a chimeric protein antibody composed of variable and constant regions from mouse and human sources, respectively, while panitumumab and nimotuzumab are fully humanized EGFR antibodies. To date, the Food and Drug Administration has approved EGFR-targeted mAbs for use in advanced colorectal cancer, gliomas, and head and neck tumors. Patients receive the drugs intravenously.
On the other hand, EGFR-specific small molecule inhibitors such as erlotinib (Tarceva) and gefitinib (Iressa), which are taken orally, translocate across the plasma membrane and interact with the cytoplasmic domain of EGFR. Small molecule inhibitors belong to a class of therapeutics called tyrosine kinase inhibitors or TKIs. In the cell, TKIs compete with ATP to bind the catalytic domain of the kinase, which in turn inhibits EGFR autophosphorylation and downstream signaling, including cell proliferation and survival. TKIs have been approved for treatment of advanced NSCLC and pancreatic carcinoma.
Small molecule inhibitors are thought to be less specific than mAbs since they can potentially target any tyrosine kinase, diluting the therapeutic effect on the target of interest. To increase the specificity and improve clinical effectiveness of TKIs, researchers have also developed bi-specific inhibitors such as lapatinib that target EGFR/ErbB2. However, the overall clinical effectiveness of these therapies and their correlation to EGFR expression is not clear. EGFR inhibitors are effective in only a small subset of patients, despite high levels of EGFR expression. Some cancers appear to acquire resistance to EGFR inhibitors, and multiple mechanisms seem to underlie the lack of sensitivity to the targeted therapies, including mutations in the EGFR gene itself, as well as in down-stream effectors such as RAS, RAF, and AKT that appear to be associated with differential clinical outcomes.
Impact of Mutations in EGFR and Downstream Signaling Pathways
The EGFR gene is present on chromosome 7p11.2 and has 28 exons coding for a transmembrane receptor protein of 464 amino acids. Within EGFR, exons 5–7 and 13–16 code for the ligand binding domain while exons 18–24 code for the TK domain. Autophosphorylation occurs in the region encoded by exons 25–28.
Although mutations can occur anywhere within the TK domain, a significant set of EGFR mutations in lung cancer that are associated with objective response to single agent TKI therapy are observed in exons 18–21. The most frequent of these are in-frame deletions in exon 19 that occur in approximately 45% of cases, followed by point mutations in exon 21, in 40–45% of cases. While more than 20 different deletions are observed in exon 19, L858R in exon 21 is the most common point mutation detected (Figure 2). Greater response to TKIs also correlates with EGFR amplification that frequently coexists with EGFR mutations and is more common in gefitinib-sensitive NSCLC with increased expression of ErbB3.
Figure 2
Spectrum of EGFR Mutations
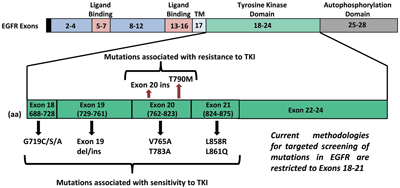
Researchers have also observed resistance to anti-EGFR therapies in a significant number of cancers, altering the clinical impact of anti-EGFR treatments. De novo mutations are known to occur within EGFR that constitutively turn on the receptor, allowing the tyrosine kinase to work much better than normal and overcoming the effects of the TKI. While the most important of these is T790M, a point mutation in exon 20 that accounts for about 50% of cases, insertional mutations in exon 20, which occur in about 5% of cases, have also been associated with TKI resistance.
Primary resistance to EGFR TKIs is mostly due to the presence of wild-type EGFR, since these tumors harbor mutations in other genes downstream of EGFR such as KRAS and BRAF that may play a role in predicting clinical response to anti-EGFR therapies. Mutations in KRAS at codons 12 and 13 occur in about 15–50% of NSCLC patients, while BRAF mutations are detected in 1–2% of lung cancer patients. KRAS and EGFR mutations appear to be mutually exclusive in NSCLC, with EGFR mutations occurring in non-smokers and KRAS mutations in smokers. Exclusivity is also observed in the case of BRAF and EGFR mutations. This makes it difficult to evaluate whether the observed resistance to EGFR-targeted TKIs is due to the presence of mutated KRAS or BRAF or the absence of mutated EGFR.
Inactivation of PTEN is observed in several cancers, including colon, breast, brain, gastric, and lung, which results in activation of the PI3K/AKT pathway and subsequent degradation of EGFR that generates insensitivity to anti-EGFR therapies. EML4-ALK fusions are a rarer abnormality only detected in 3–13% of lung adenocarcinomas, mutually exclusive of EGFR status. Lung cancers positive for these fusions are resistant to EGFR-targeted TKIs but sensitive to ALK TKIs such as crizotinib (also known as Xalkori).
Acquired or secondary resistance to EGFR-targeted TKI therapies also includes secondary mutations in EGFR, particularly the T790M that is observed post gefitinib treatment and is thought to occur due to selection pressure. Similarly, mutations in PIK3CA and MET are also believed to be associated with acquired resistance. PIK3CA mutations occur in approximately 5% of EGFR mutant lung cancers, while amplification of MET is detected in up to 20% of lung cancer specimens that developed acquired resistance to gefitinib or erlotinib. The data suggests that EGFR TKI treatment may select for preexisting cells with MET amplification during the acquisition of EGFR TKI resistance.
Testing for Mutations in the EGFR Pathway
Clearly, the therapeutic implications of EGFR pathway mutations are substantial. Laboratory studies to identify the mutations are therefore integral to evaluating the efficacy of anti-EGFR therapies designed to manage and treat patients with positive test results.
Traditional methods for detecting EGFR mutations involved screening by direct DNA sequencing of the tumor tissue. Sanger sequencing technology is available in most molecular diagnostic laboratories, and it has the singular advantage of detecting alterations across a gene, including novel variants. However, the sensitivity of this method is low. In a heterogeneous tumor, there must be at least 10–30% mutant DNA in the background of normal DNA. Mutation screening by sequencing is also time- and labor-intensive, making the test costly.
Recent methodologies have therefore focused on targeted screening of mutations to achieve more rapid, robust, and sensitive tests. Molecular diagnostic laboratories currently use a variety of methods, including amplification refractory mutation system, pyrosequencing, smart amplification process, high-resolution melting analysis, and restriction fragment length polymorphism, to name a few. These methods all distinguish between mutant and wild-type DNA within the region of interest. In contrast to direct sequencing, the limit of detection for targeted analysis is ~1–5% mutant DNA in the background of normal DNA. The disadvantage, of course, is that the assay will only detect those specific mutations targeted by the assay.
Laboratories would be well advised to choose a method after considering a number of factors, including availability of instrumentation, reagents required, and other preferences and practices in the lab. However, by far the most important consideration in choice of methodology is the amount of sample available for testing. More often than not, a very small amount of tumor is available. Furthermore, clinicians need the test results quickly to evaluate drug sensitivity or resistance, so the turnaround time must be as short as possible. In many cases, depending upon the technology and platform used, establishing a viable cost-benefit ratio also becomes a challenge. In sum, these considerations generally point to targeted screening of EGFR mutations.
Today, most laboratories use formalin-fixed, paraffin-embedded (FFPE) tissue to test for EGFR mutations. A few also use frozen tumor tissue. However, screening FFPE samples poses significant challenges, including successfully extracting the DNA, interferences from the fixatives used for embedding tissue, and most importantly, obtaining shorter amplicons for effective analysis, particularly if a PCR-based methodology is used.
Alternate sample types such as fine needle aspirates and pleural effusions are currently being evaluated as viable options to enable quicker, easier diagnosis of malignancy. Micro-dissection of the tumor prior to testing is also helpful as it effectively enriches the sample, thereby increasing sensitivity.
Strategies for Therapy
Understanding the efficacy of anti-EGFR treatments, as well as how efficacy correlates to mutations within the EGFR, RAS, BRAF, and other pathways, as well as with crosstalk to the EGFR pathway, has proven to be challenging. In a majority of cancers, single therapies are either short-lived or completely ineffective. Some of the factors that may contribute to these observations are the presence of mutations in multiple pathways and the induction of mutations in non-targeted pathways that have a domino effect, impacting efficacy of the targeted therapy by feedback inhibition (acquired resistance).
Given these shortcomings in targeted therapies, researchers are also evaluating combinatorial therapies, such as sorafenib (targets RAF) with erlotinib, for EGFR mutations and the downstream signaling pathways in NSCLC and glioblastoma. Similarly in patients with both EGFR and PIK3CA mutations, clinicians might consider combination therapy with an EGFR TKI and a PI3K inhibitor. Solving this problem will require algorithms that facilitate testing for mutations, not just in EGFR, but also in molecules downstream (RAS, RAF, AKT, and PIK3CA, etc.). This information might allow clinicians to predict patients' clinical response to standard therapies, particularly for combinatorial approaches that in theory appear to be the most effective.
Many questions need to be answered in order to effectively treat patients with defects in the EGFR family of pathways. Should patients be tested for de novo mutations sequentially or simultaneously in EGFR/RAS/RAF? Assuming they present with the same mutation profile, should patients' metastatic tumors be treated similarly to their primary tumor, or should the metastatic tumor be tested prior to treatment? Should patients receive personalized treatment based on the mutation profile that involves combinatorial therapies not evaluated before? Should pharmaceutical companies actively evaluate multi-arm combinatorial therapy clinical trials targeting EGFR pathways similar to those in triple-negative/triple-positive breast cancer?
To determine the most appropriate treatment for an individual, the American Society for Clinical Oncology recommends that patients with advanced NSCLC who have not previously had chemotherapy or an EGFR TKI drug have their tumors tested for EGFR mutations before therapy is administered. Given the acquired resistance to anti-EGFR therapies observed in tumors, ideally laboratories would test the primary, as well as recurrent metastatic tumors to ensure effective clinical response.
Clinicians and molecular testing laboratories will need to keep up-to-date with the growing body of literature on the mechanisms involved in modulation of signaling pathways such as EGFR in cancer, as well as its sensitivity and resistance to currently available treatments. Currently, there is no right or wrong strategy, only an appropriate one that can be personalized to benefit the patient.
SUGGESTED READING
Cheng L, Alexander RE, Maclennan GT, et al. Molecular pathology of lung cancer: Key to personalized medicine. Mod Pathol 2012;25:347–69.
Ellison G, Zhu G, Moulis A, et al. EGFR mutation testing in lung cancer: A review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol 2013;66:79–89.
Lin L, Bivona TG. Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in NSCLC patients. Chemother Res Pract 2012;2012:817297.
Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin Cancer Res 2006;12:5268–72.
Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169–81.

Honey V. Reddi, PhD, is laboratory director of molecular genetics at Prevention Genetics, Marshfield, Wisc.
Email: [email protected]
Disclosure: The author has nothing to disclose.