Summary
DOI: 10.1373/clinchem.2015.246033
A 51-year-old woman consulting within the framework of investigation for abdominal discomfort, nausea, and vomiting underwent a computed tomography examination that revealed a well-delimited right adrenal heterogeneous mass measuring 8.2 × 8.3 cm with a native density of 40 Hounsfield units (HU)8. An 18F-deoxyglucose positron emission tomography scan showed a hypercaptation on the adrenal tumor of 7.4 SUVmax. Apart from these symptoms, the patient had no other complaint. Familial history was unremarkable. On examination, the patient was in good physical condition, arterial blood pressure was 136/85 mmHg, heart rate at 74 bpm.
Student Discussion
Student Discussion Document (pdf)
Karim Abid,1† Katayoun Afshar,2† Enzo Fontana,3 Julien Ducry,3 Samuel Rotman,4 Edouard Stauffer,5 Florence Fellmann,6 Oliver Tschopp,7 Zahurul A. Bhuiyan,2† and Eric Grouzmann1*†
1Département des Laboratoires, Laboratoire des Catécholamines et Peptides, University Hospital of Lausanne, Lausanne, Switzerland; 2Laboratoire de Diagnostic Moléculaire, Service de Génétique Médicale, University Hospital of Lausanne, Lausanne, Switzerland; 3Service d’Endocrinologie et Diabétologie de l’Hôpital Cantonal de Fribourg, Villars-sur-Glȃne, Switzerland; 4Institute of Pathology, University Hospital of Lausanne, Lausanne, Switzerland; 5Pathologie, Promed Laboratoire Médical SA, Fribourg, Switzerland; 6Service de Génétique Médicale, University Hospital of Lausanne, Lausanne, Switzerland; 7Division of Endocrinology, Diabetes and Metabolism, University Hospital of Zurich, Zurich, Switzerland.
†Karim Abid and Katayoun Afshar contributed equally to this article and Zahurul A. Bhuiyan and Eric Grouzmann shared senior authorship.
* Address correspondence to this author at: Laboratoire des Catécholamines et Peptides, Département des Laboratoires, CHU Vaudois, Hôpital Nestlé, Av Pierre Decker, 1011, Lausanne, Switzerland. Fax +41-21-314-7835; e-mail [email protected].
8Nonstandard abbreviations: HU, Hounsfield units; DBH, dopamine _-hydroxylase; PNMT, phenylethanolamine-N-methyl transferase.
9Human genes: TH, tyrosine hydroxylase; DBH, dopamine beta-hydroxylase; PNMT, phenylethanolamine N-methyltransferase; SDHB, succinate dehydrogenase complex iron sulfur subunit B; SDHD, succinate dehydrogenase complex subunit D; VHL, von Hippel-Lindau tumor suppressor; RET, ret proto-oncogene.
Case Description
A 51-year-old woman consulting within the framework of investigation for abdominal discomfort, nausea, and vomiting underwent a computed tomography examination that revealed a well-delimited right adrenal heterogeneous mass measuring 8.2 × 8.3 cm with a native density of 40 Hounsfield units (HU)8. An 18F-deoxyglucose positron emission tomography scan showed a hypercaptation on the adrenal tumor of 7.4 SUVmax. Apart from these symptoms, the patient had no other complaint. Familial history was unremarkable. On examination, the patient was in good physical condition, arterial blood pressure was 136/85 mmHg, heart rate at 74 bpm.
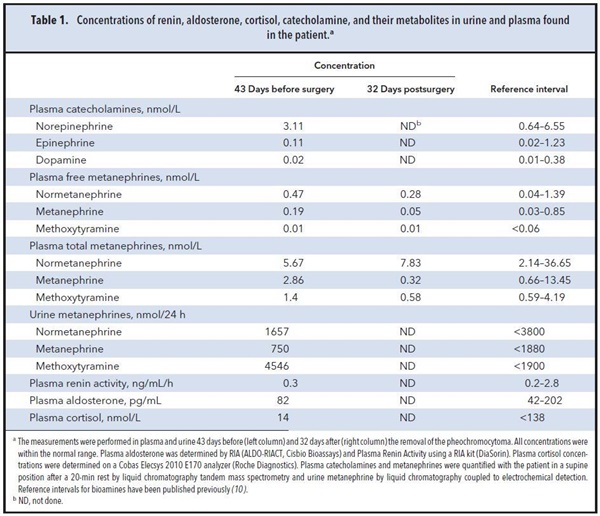
The measurement of plasma renin activity and aldosterone and the results of a 1-mg overnight dexamethasone suppression test ruled out primary hyperaldosteronism and Cushing syndrome. Catecholamine and metanephrine concentrations in multiple blood and urine samples were consistently within the reference interval and not compatible with the diagnosis of a pheochromocytoma (Table 1).
Because the adrenal mass was not hormonally active, its surgical resection was performed without preoperative care. The removal of a mass weighing 356 g in the right adrenal gland was performed by open laparotomy.
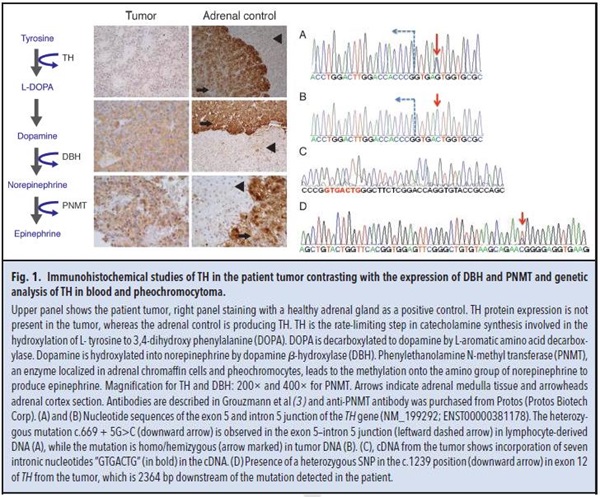
Histopathological analysis of tissue sections of the adrenal mass yielded an unexpected result: a pheochromocytoma was diagnosed on the basis of typical well-arranged nests called zellballen. This encapsulated adrenal tumor exhibits a low proliferation index (MIB-1, 1%–2%; 1 mitosis/high power field) without vascular invasion. Immunohistochemistry of the tumor sections revealed its neuroendocrine feature, with cells highly expressing CD56, chromogranin A, NSE, synaptophysin, and vimentin. Unfortunately, blood samples obtained before surgery were collected on heparin-coated tubes precluding serum chromogranin A assay. Because the unusual presentation of a “non–catecholamine-secreting pheochromocytoma” was in complete contradiction with the biochemical feature expected for these tumors, we studied the intratumoral protein expression of the main enzymes involved in catecholamine synthesis by means of immunohistochemistry on paraffin-embedded sections of the tumor biopsy. Tyrosine hydroxylase (TH) was not expressed in tumor tissue compared to normal adrenal gland medulla (Fig. 1 upper panel). Dopamine β-hydroxylase (DBH) and phenylethanolamine-N-methyl transferase (PNMT) protein expressions were lower in the tumor than in the adrenal medulla control, but the cells were clearly stained (Fig. 1 middle and lower panels). The TH (tyrosine hydroxylase)9 gene expression was approximately 20-fold lower in this tumor than in the adrenal pheochromocytoma (P = 0.016), whereas expressions for DBH (dopamine beta-hydroxylase) and PNMT (phenylethanolamine N-methyltransferase) were unchanged (P = 0.29 and P =0.67, respectively). Routine blood DNA analyses of the most common genes associated with a familial risk for a pheochromocytoma, SDHB (succinate dehydrogenase complex iron sulfur subunit B), SDHD (succinate dehydrogenase complex subunit D), VHL (von Hippel-Lindau tumor suppressor), and RET (ret protooncogene), were performed and revealed no mutations.
To investigate the molecular mechanism associated with the absence of expression of TH, the sequence of the coding exons and the exon–intron boundaries of TH were analyzed using the DNA from both blood lymphocytes and tumoral tissue of the patient (Fig. 1A and B). Sequencing of TH in the tumor tissue uncovered a homo/hemizygous mutation, c.669 + 5 G>C, in the exon 5/intron 5 junction of TH (Fig. 1B). The genomic location of this mutation is at Chr11: 2189316 (http://exac.broadinstitute.org/). This mutation has also been detected in the DNA from the lymphocytes, but only in the heterozygous state. This mutation was located in the conserved region for RNA splicing (donor site), at the 3'-junction of exon and intron 5, and was likely to affect RNA splicing efficiency.
Questions to Consider
- How can one define an adrenal pheochromocytoma that is unable to secrete catecholamines?
- What alternative may be proposed to monitor a non-catecholamine-secreting pheochromocytoma?
- How can one monitor for a possible relapse of a pheochromocytoma for this patient?
- Are special measures needed preoperatively before removal of a non-catecholamineproducing adrenal pheochromocytoma?
Final Publication and Comments
The final published version with discussion and comments from the experts appears
in the July 2016 issue of Clinical Chemistry, approximately 3-4 weeks after the Student Discussion is posted.
Educational Centers
If you are associated with an educational center and would like to receive the cases and
questions 3-4 weeks in advance of publication, please email [email protected].
AACC is pleased to allow free reproduction and distribution of this Clinical Case
Study for personal or classroom discussion use. When photocopying, please make sure
the DOI and copyright notice appear on each copy.
DOI: 10.1373/clinchem.2015.246033
Copyright © 2016 American Association for Clinical Chemistry