Over the last 50 years there have been great improvements in managing sickle cell disease (SCD) in the United States. In the 1970s, the median age of death for a person born with SCD was 14; today, the median age of death is in the late 40s.
Until 2017, treatment options for SCD largely consisted of red blood cell (RBC) transfusion and hydroxyurea (2,3). The Food and Drug Administration (FDA) recently approved three additional medications, increasing the available options for disease management to five. These include inflammation reducers such as L-glutamine; a new monoclonal therapy, crizanlizumab; and voxelotor (formerly GBT440), a novel drug that inhibits the hemoglobin polymerization process that causes RBC sickling (4−6).
In addition to these treatments, hematopoietic stem cell transplantation and gene therapies are potential curative treatments (7,8). This article provides an update on the current treatment options available and the current laboratory methods used to monitor patients with SCD.
Background On SCD
SCD is the most common inherited blood disorder in the United States, affecting more than 100,000 Americans and millions of people worldwide (1). SCD occurs as a result of changes to the protein hemoglobin, a major carrier of oxygen in the blood. The majority of adult hemoglobin is composed of hemoglobin A (HbA), with small amounts of hemoglobin A2 (HbA2) and hemoglobin F (HbF) (Table 1). SCD is an autosomal recessive disease caused by a point mutation in the β-globin gene that produces an abnormal hemoglobin S (HbS) instead of wild-type HbA.
These mutations cause the production of dysfunctional hemoglobin that polymerizes in the setting of hypoxia into stiff rods, causing RBCs to take on a sickled shape (sickling). Sickled RBCs are unable to move through small blood vessels (vaso-occlusion) and are easily destroyed via hemolysis (1). As a result, tissue beyond the area of vaso-occlusion cannot receive oxygen and dies (ischemia), leading to extreme pain. Episodes of vaso-occlusion are known as pain crises and are the most common cause of morbidity in SCD.
SCD occurs when an individual inherits either two copies of HbS (HbSS) or one copy of HbS and a second misfunctioning β-globin gene, such as hemoglobin C (HbC), hemoglobin D-Los Angeles/Punjab (HbD-Los Angeles/Punjab), hemoglobin E (HbE), hemoglobin O-Arab (-Arab), or β-thalassemia, which is caused by either a mutation in one or more of the β-globin genes or deletion of an entire β-globin gene (Table 1).
Understanding SCD Treatment Options
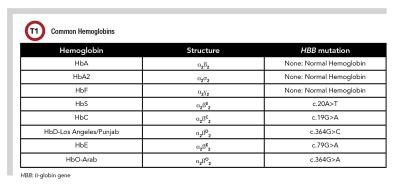
There are currently five available treatment options for managing SCD: RBC transfusions (simple or exchange); hydroxyurea (approved by FDA in 1998); L-glutamine (approved in 2017); crizanlizumab (approved in 2019); and voxelotor (approved in 2019) (2−6). These treatments work by targeting either the initial event and/or areas of downstream dysfunction (Figure 1).
RBC Transfusion
RBC transfusions are used for prevention and management of SCD symptoms, including acute chest syndrome, recurrent vaso-occlusive pain crises, acute stroke treatment, and stroke prophylaxis. The goal of RBC exchange is to reduce the relative percentage of HbS (HbS%) and to increase oxygen delivery to the tissues, thereby preventing sickling events and subsequent vaso-occlusion, hemolysis, and ischemia (2).
Hydroxyurea
In 1998, hydroxyurea was the first FDA-approved medication for the treatment of SCD after it was proven to reduce frequency of pain crisis in adults (3). Hydroxyurea is an inhibitor of ribonucleotide reductase and, similar to RBC transfusions, reduces the relative HbS%. Hydroxyurea does this by increasing the production of β-globulin, which in turn increases the percentage of HbF (HbF%) in the blood.
HbF is composed of two α and two γ subunits and is unaffected by the HbS mutation (Table 1). Increasing the HbF% in the blood decreases the HbS% and substitutes a hemoglobin that does not polymerize. Hydroxyurea also inhibits cell replication, reducing white cell and platelet counts. This reduces chronic inflammation and subsequently reduces the risk of vaso-occlusion.
Hydroxyurea is a tablet taken daily and remained the sole medication approved for the treatment of SCD for almost 2 decades.
L-glutamine
L-glutamine was approved by FDA for the treatment of SCD when it was shown to reduce frequency of pain crises both alone and in combination with hydroxyurea (4). L-glutamine is an amino acid necessary for the synthesis of nicotinamide adenine dinucleotide (NAD). Higher levels of NAD have been proposed to reduce oxidative stress resulting from ischemia reperfusion reactions in SCD, which would subsequently reduce chronic inflammation. L-glutamine is a pharmaceutical-grade powder that is mixed in a beverage or food and taken twice daily.
Crizanlizumab
Crizanlizumab is a monoclonal antibody for p-selectin, a cell adhesion molecule found on platelets and endothelial cells (5). It is taken as a monthly intravenous infusion and has been shown to reduce episodes of pain crises alone and in combination with hydroxyurea. By blocking p-selectin it reduces cell adhesion to the endothelium, subsequently reducing vaso-occlusive events (5).
Voxelotor
Voxelotor is a small molecule that reversibly binds to one α-globin chain within the hemoglobin tetramer and increases the hemoglobin affinity to oxygen (6). This increase in affinity is thought to prevent polymerization and reduce sickling events and subsequent hemolysis.
Voxelotor has been found to increase hemoglobin levels both alone and in combination with hydroxyurea. It was approved in late 2019 for the treatment of SCD in adults and pediatric patients >12 years of age, and in 2021 this approval was extended to patients >4 years of age. It is a tablet taken once a day for patients >12 and a tablet for oral suspension for patients aged 4−11 (9).
Combination Therapy
L-glutamine, crizanlizumab, and voxelotor were all initially studied and shown to be beneficial both alone and in combination with hydroxyurea; however, no other combinations of therapies have been examined. Future studies are needed to determine if other combinations of approved therapeutics may also be beneficial in the treatment of sickle cell disease.
SCD Treatments With Curative Intent
Hematopoietic Stem Cell Treatment
Allogenic stem cell transplants have been shown to cure SCD by replacing the patient’s native bone marrow with a transplanted marrow that will produce RBCs with HbA instead of HbS. A study of 1,000 patients who underwent matched related donor transplants showed a 5-year survival of 92.9% and event-free survival of 91.4%.
While hematopoietic stem cell transplant is a possible curative therapy, there are currently significant barriers to using it for most patients with SCD. An estimated 18% of people living with SCD have a possible matched related donor, and only an additional 18% of people living with SCD have a possible donor in the registry. The rates of event-free survival are lower for patients whose transplant was from a nonrelated matched donor from the bone marrow registry (7).
Gene Therapy
Gene therapy may be a possible curative treatment option for those who do not have a possible matched related donor for stem cell transplantation. Gene therapy works by taking the patient’s own stem cells, modifying them, and then replacing the patient’s native bone marrow with the modified cells.
Multiple modification methods and targets for modification are currently being studied, and it remains to be seen which methods will prove superior. Additionally, researchers have yet to determine whether the effects of gene modification will last long enough for this to serve as a curative treatment. However, studies haven’t ruled out the potential for gene therapy to offer a curative treatment for some SCD patients either (8).
Laboratory Methods For Monitoring And Managing Patients
There are numerous methods available to diagnose hemoglobinopathies. The first is hemoglobin fractionation and consists of separating hemoglobin species based on charge and/or size. Hemoglobin fractionation can be performed by high-performance liquid chromatography (HPLC) methods, capillary electrophoresis (CZE) methods, and gel electrophoresis methods (alkaline and acid gels and isoelectric focusing [IEF] gels).
HPLC separates hemoglobin fractions based on charge, whereas with CZE and gel methods, separation is based on both size and charge. HPLC and CZE are high resolution quantitative techniques. Gel methods, on the other hand, are low resolution compared to HPLC and CZE, although they can be semiquantitative.
Mass spectrometry and molecular methods (sequencing and deletion/duplication) are extremely powerful techniques for diagnosing hemoglobinopathies and thalassemia, particularly those that are challenging to diagnose with HPLC, CZE, and gel methods. However, these methods are for qualitative identification and do not quantify hemoglobin species.
Patients with SCD have historically been managed by RBC transfusions and/or hydroxyurea. Managing and monitoring patients on these treatments requires discrimination and accurate quantitation of the various hemoglobin species; therefore, HPLC or CZE are the preferred methods for following
these patients.
Gel methods, molecular genetics, and mass spectrometry methods are powerful tools for diagnosis but are not as well-suited for managing SCD. The 2021-B College of American Pathologists (CAP) hemoglobinopathy peer evaluation survey showed that 95% of respondents quantified hemoglobin species via HPLC or
CZE methods.
Monitoring RBC Transfusion
Typically, quantitation of HbS% is completed prior to transfusion (pretransfusion) to calculate the units of RBCs to be transfused. A posttransfusion HbS% measurement is used to determine efficacy of treatment. In addition to hemoglobin fractionation, iron overload is a concern for patients undergoing transfusions and iron and ferritin should be monitored. Alloantibodies should also be monitored to limit the potential of transfusion reactions, especially after repeated transfusions.
Managing Hydroxyurea Dosage and Monitoring Compliance
For patients treated with hydroxyurea, HbF% is used to monitor dosage and compliance. In addition to hemoglobin fractionation, patients treated with hydroxyurea are monitored with serial CBC measurements to determine efficacy of treatment and guide dosing changes. As hydroxyurea doses are increased, HbF increases and inflammation markers decrease—and at high doses, neutrophil populations decrease and general leukopenia eventually sets in.
Typically, hydroxyurea dosing is increased until the patient’s maximally tolerated dose (MTD) is reached. CBC parameters such as white blood cell and neutrophil counts are monitored to determine this point and to ensure that patients have sufficient leukocytes. As cells with HbF are typically larger than other RBCs, the mean corpuscular volume may also be used to monitor treatment efficacy.
Monitoring Stem Cell Transplants
Hemoglobin quantification is often performed on patients who undergo stem cell transplant to assess the health of the graft.
Crizanlizumab
Crizanlizumab is a monoclonal antibody treatment approved in 2019. As crizanlizumab is delivered through an infusion, most monitoring is acute and centered around infusion-related reactions such as pain, gastrointestinal upset, and fever. It is also important to note that p-selectin is displayed on platelet surfaces and there have been reports of platelet clumping after administration, particularly for collections into EDTA-containing collection devices.
Platelet clumping is a postcollection phenomenon and does not reflect an in vivo condition; however, it can falsely reduce platelet counts on automated analyzers. Because of this, platelet counts from tubes with citrate or heparin anticoagulant (which do not show this effect) may sometimes be used for monitoring in this patient population.
Interference Of Voxelotor With HbS% Quantitation
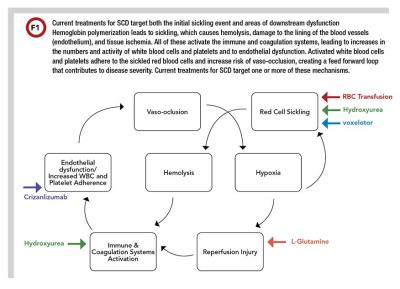
In January 2018, a patient presented to the emergency department at Vanderbilt University Medical Center in sickle cell crisis. An emergent RBC exchange transfusion was scheduled and pre-exchange HbS% quantitation ordered. The patient had a reported diagnosis of genotype HbSS. Our laboratory noted an unusual chromatogram and paged the attending clinical chemists for a consult. Instead of a single distinct peak in the HbS peak region, there were two overlapping peaks that spanned the HbD and HbS windows, giving the appearance of a “split peak” (Figure 2b).
Interestingly, a review of our patient’s previous chromatogram showed a well resolved HbS peak, consistent with his HbSS genotype (Figure 2a) (10). We discussed our findings with the clinical team and the patient ultimately received 10 units of RBCs. Our patient presented 2 weeks after his RBC exchange for follow up. In addition to the “split peak,” the hemoglobin fractionation chromatogram exhibited two additional distinct peaks near HbF and HbA (Figure 2c) (10).
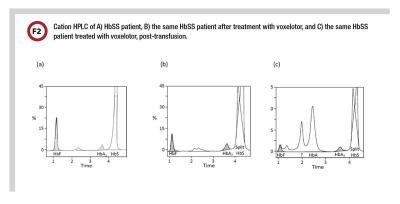
Upon further investigation, we discovered that the patient had recently been enrolled in a phase 3 clinical trial for voxelotor (NCT03036812) and was not taking the placebo. Given that the α-globin chain is not specific to HbS, we suspected that the additional peaks could be due to other voxelotor-hemoglobin complexes. Hemoglobin fractionation by CZE on the posttransfusion sample also demonstrated a “split HbS” peak and two additional peaks (Figure 3).
With our colleagues, we conducted qualitative MALDI mass spectrometry studies that confirmed the presence of voxelotor-hemoglobin α chains in our patient’s sample (10). Further studies demonstrated that patients treated with voxelotor also exhibit a “split HbS” peak and additional peaks when their hemoglobin fractionation profiles are measured via CZE (Figure 3).
To our knowledge, this was the first report that patients treated with voxelotor exhibited markedly altered HPLC and CZE chromatograms and additional peaks in IEF (10).
To assess how voxelotor affects accurate quantitation, we performed in vitro spiking experiments to mimic voxelotor treatment and quantified the impact on HPLC, CZE, and acid and alkaline gels (11). These studies demonstrated that the formation of voxelotor-hemoglobin species affects accurate quantitation of multiple different hemoglobin species. The 2021-B CAP hemoglobinopathy peer evaluation survey showed that approximately 87% of laboratory respondents quantified hemoglobin species via HPLC, CZE, or gel methods that were evaluated in this study and would be unable to resolve this interference. The remaining HPLC and CZE methods were not evaluated in this study.
The Role of Clinical Laboratories in Patient Management
Clinical laboratories should take an active role in ensuring that SCD patients get effective treatment. In the case of voxelotor specifically, laboratorians who are at centers where patients are treated with this drug should be aware of how to recognize its interference and the impact it can have on reporting results.
One potential way of communicating this interference to clinicians is the addition of a comment in the report notifying clinicians. One laboratory also has reported creating an automated order inquiry for quantitative hemoglobin fractionation that asks the clinician if the patient is taking voxelotor (12). Close communication between hematologists, transfusion medicine physicians, and laboratorians is crucial to successfully managing these patients.
A key question is whether the HbS-voxelotor complexes should be considered clinically equivalent to HbS for the purposes of transfusion or medication management, or if they should be treated as two distinct species, one sickling and one nonsickling. This will have implications for how results are reported, which currently available testing methods should be used, and if a separate field is needed to account for Hb-voxelotor complexes, including HbS-voxelotor.
The ability to accurately detect Hb-voxelotor complexes is exciting due to the potential for monitoring patient compliance, dose titration, and quantitative measurements that can be linked to patient outcomes. However, clinical consensus and guidance are still needed on how to report hemoglobin species in patients treated with voxelotor (11)
Acknowledgements: The authors would like to thank Jennifer Colby, PhD, DABCC, FADLM, for her critical review of the manuscript.
References
1. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med 2017;376:1561−73
2. Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv 2020;4:327−55.
3. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317−22.
4. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of L-glutamine in sickle cell disease. N Engl J Med 2018;379:226-35.
5.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 2017;376:429−39.
6. Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med 2019;381:509−19.
7. Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017;129:1548−56.
8. Abraham AA and Tisdale JF. Gene therapy for sickle cell disease: Moving from the bench to the bedside. Blood 2021;138:932-41.
9. Oxbryta (voxelotor) package insert. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213137s006lbl.pdf (Accessed May 2022).
10. Rutherford NJ, Thoren KL, Shajani-Yi Z, et al. Voxelotor (GBT440) produces interference in measurements of hemoglobin S. Clin Chim Acta 2018;482:57−.
11. Rutherford-Parker NJ, Campbell ST, Colby JM, et al. Voxelotor treatment interferes with quantitative and qualitative hemoglobin variant analysis in multiple sickle cell disease genotypes. Am J Clin Pathol 2020;154:627−34.
12. Godbey EA, Anderson MR, M Bachmann L, et al. How do we monitor hemoglobin S in patients who undergo red blood cell exchange and take voxelotor? Transfusion 2021;61:1680−3.
Sean T. Campbell, PhD, DABCC, FADLM, is the interim director of chemistry and immunology at the Montefiore Medical Center in New York City. +Email: [email protected]
Susanna Curtis MD, PhD, is the assistant director of sickle cell disease at Mount Sinai Hospital and assistant professor at Icahn School of Medicine at Mount Sinai in New York.. +Email: [email protected]
Zahra Shajani-Yi, PhD, DABCC, FADLM, NRCC-CC, is technical director of clinical chemistry at Labcorp’s San Diego regional laboratory in California and national co-discipline director of routine and esoteric immunoassays. This work took place while she was medical director of esoteric chemistry at Vanderbilt University Medical Center and assistant professor at Vanderbilt University School of Medicine in Nashville. +Email: [email protected]