
Next-generation sequencing (NGS)-based molecular tests have revolutionized the practice of medicine with the ability to personalize diagnosis, risk assessment, and treatment of patients with cancer and non-neoplastic disorders. Given the vast amounts of quantitative and complex sequencing data generated by high-throughput sequencers, clinical laboratories rely on resource-intensive data processing pipelines to analyze data and identify genetic alterations of clinical relevance.
Bioinformatics, specifically in the context of genomics and molecular pathology, uses computational, mathematical, and statistical tools to collect, organize, and analyze large and complex genetic sequencing data and related biological data. A set of bioinformatics algorithms, when executed in a predefined sequence to process NGS data, is collectively referred to as a bioinformatics pipeline (1).
Clinical molecular laboratories performing NGS-based assays have as an implementation choice one or more bioinformatics pipelines, either custom-developed by the laboratory or provided by the sequencing platform or a third-party vendor. A bioinformatics pipeline typically depends on the availability of several resources, including adequate storage, computer units, network connectivity, and appropriate software execution environment. Ensuring consistent, on-demand access to these resources presents several challenges in clinical laboratories.
This article will discuss some important practical considerations for laboratory directors and bioinformatics personnel when developing NGS-based bioinformatics resources for a clinical laboratory. This short review is not a comprehensive guide for all aspects of bioinformatics resource development. Readers should consult the references for additional details.
The Bioinformatics Workflow in Clinical Laboratories
An emerging sub-specialty in laboratory medicine, clinical bioinformatics focuses on the application of bioinformatics principles, methods, and software tools to analyze, integrate, and understand biologic and healthcare data in a clinical setting (2). Clinical bioinformatics has several applications in a clinical molecular laboratory offering NGS-based testing.
NGS generates several million to billion short-read sequences of the DNA and RNA isolated from a sample. In contrast to traditional Sanger sequencing, with read lengths of 500-900 base pairs (bp), short reads of NGS range in size from 75 to 300 bp depending on the application and sequencing chemistry. Newer NGS technologies such as those from PacBio, Nanopore, and 10x Genomics enable longer read sequences in excess of 10 kilobases (3).
NGS also sequences RNA molecules by converting them to complementary DNA (cDNA) molecules using reverse-transcriptase polymerase chain reaction. In addition to the sequence itself and unlike Sanger sequencing, the high-throughput nature of NGS provides quantitative information (depth of coverage) due to the high level of sequence redundancy at a locus. This property of NGS data enables laboratories to identify a vast repertoire of genetic alterations from a single NGS run on a sample using different bioinformatics algorithms (4) (Figure 1).
Click to View Full Size Image
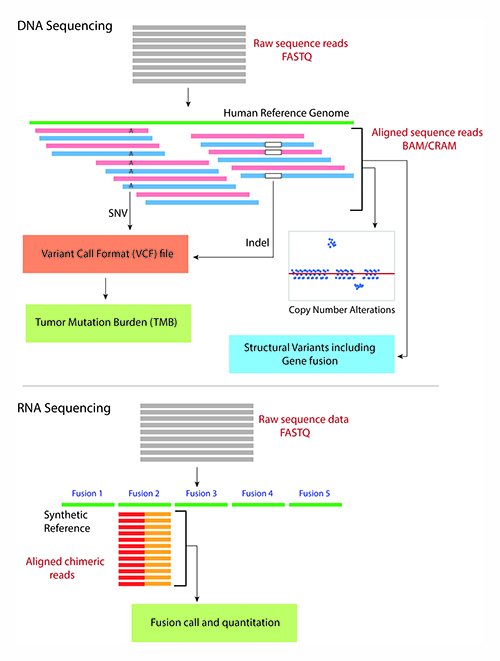
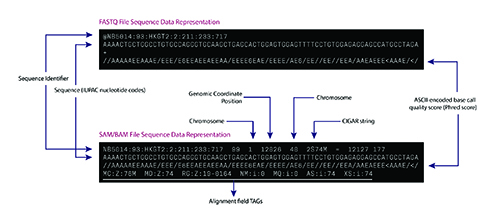
The bioinformatics pipeline for a typical DNA sequencing strategy involves aligning the raw sequence reads from a FASTQ or unaligned BAM (uBAM) file against the human reference genome. The FASTQ and uBAM file formats store short sequences as plain text with metadata about each short sequence such as base quality score and read identifiers (Figure 2a).
The sequence alignment process assigns a genome positional context to the short reads in the reference genome and generates several metadata fields, including alignment characteristics (matches, mismatches, and gaps) in Concise Idiosyncratic Gapped Alignment Report format. The aligned sequences and the related metadata are stored in a Sequence Alignment Mapping (SAM/BAM) (Figure 2b) or CRAM file format (5). Downstream algorithms consume the BAM file to identify a range of genetic alterations, including single nucleotide variants, insertions and deletions (indels), and tumor mutation burden (4,6).
Click to View Full Size Image
Laboratories commonly estimate copy number alterations (CNA) from aligned sequencing reads by using the depth of coverage approach. More extensive and specific DNA sequencing strategies also enable identification of large structural variants (SV), including gene fusions, and microsatellite instability (6,7). In addition, a split-read alignment strategy identifies gene fusions from genomic DNA sequencing (7).
For RNA-based gene fusion detection using NGS, the bioinformatics process typically involves aligning the cDNA sequences to an artificially constructed genome containing a list of known fusion sequences. The total number of reads from the sample that align to one of the known fusion sequences can be counted to identify and quantify the gene fusion (Figure 1) (8).
The results of variant identification are stored in one of the variant call formats (VCF), including genome VCF, generic feature format, and others. These formats allow encoding quantitative information about the variant, such as variant allele fraction, depth of coverage at the variant position, and genotype quality. Given the more complex representation of CNA and large SV, including gene fusions, there is ongoing work on using alternative file formats to represent such data types appropriately (9).
Finally, the downstream bioinformatics analysis for DNA sequence variants involves queries across multiple genomic databases to extract meaningful information about gene and variant nomenclature, variant prevalence, functional impact, and assertion of clinical significance. A user interface renders and visualizes annotated DNA sequence variants, CNA, SV, and other genetic alterations (4,6). Such a user interface allows trained molecular pathologists and practitioners to interpret the clinical significance of the genetic alterations and release a comprehensive molecular report.
Additional important applications of bioinformatics in molecular laboratory operations include quality control monitoring of sequencing data across runs, identification of background sequencing noise to reduce false-positive results, validation of upgrades to the bioinformatics pipeline, and the development and validation of novel algorithms for sequence data processing and variant interpretation.
Implementing a Bioinformatics Pipeline
In order to have high confidence in the performance of NGS results, laboratories must perform a thorough validation as described in practice guidelines (1). Subsequent updates to the bio-informatics pipeline should undergo appropriate revalidation and systematic version control (See Box p. 16).
A bioinformatics pipeline and the related software interoperate closely with other devices, such as laboratory instruments, sequencing platforms, high-performance computing clusters (HPC), persistent storage resources, and other software such as laboratory information systems and electronic medical records. It is essential that the pipeline validation include such interface functions.
During the validation and implementation of bioinformatics resources in a clinical laboratory, it is crucial to ensure compliance with Federal, state and local regulations as well as specific accreditation requirements (e.g. CAP laboratory accreditation). This is particularly important for safeguarding protected health information (PHI) (4).
Detection, accurate representation, and the nomenclature of sequence variants can be challenging depending upon the variant type, sequence context, and other factors. This makes it crucial that labs understand and evaluate the region of the genome sequenced by the NGS assay for accurate clinical reporting. Appropriate automation of bioinformatics resource development and deployment in clinical production contributes to optimized test turnaround time, better productivity of the bioinformatics team, and maintainable infrastructure (10,11).
Mastering a Team Approach
Laboratory directors need to consider a multidisciplinary approach when developing bioinformatics resources. Key stakeholders should include clinical, laboratory, and hospital informatics teams, cloud and/or system architects, molecular pathologists, laboratory personnel, and the laboratory quality assurance team.
Building a robust bioinformatics infrastructure undeniably requires staff with expertise and training in bioinformatics and software engineering, strategic planning, and phased implementation, including validation and version control before clinical testing is performed. Additionally, validation automation and use of container technology can be incorporated during development or phased to a later stage based on the size of the bioinformatics team and availability of laboratory resources.
ELEMENTS OF A BIOINFORMATICS PIPELINE IN A CLINICAL LABORATORY
Bioinformatics Pipeline Implementation
Validation
The most critical requirement for implementing a bioinformatics pipeline is a proper, systematic clinical validation in the context of the entire next-generation sequencing (NGS) assay (1,12). Laboratories should determine a pipeline’s performance characteristics based on the types of variants the NGS test intends to detect and should consider the sample matrix, such as fresh tissue, peripheral blood, or formalin-fixed paraffin-embedded tissue.
A clinical laboratory, with the assistance of a bioinformatics professional or team, reviews, understands, and documents each component of the pipeline, the data dependencies, input/output constraints, and develops mechanisms to alert for unexpected errors. Command-line parameters for each component of the pipeline and their settings should be documented and locked before validating the pipeline along with an appropriate minimum number of variants, based on desired confidence and reliability, for each variant type that will be part of the validation cohort (1,12).
Version Control
Laboratories can enforce version control using software frameworks such as git, mercurial, and source control, among others. These tools enable not only systematic management of the pipeline source code but also collaborative development by a team of bioinformatics and software engineers. Version control of the pipeline should include semantic versioning of the deployed instance of a pipeline as a whole. Every deployment, including an update to the production pipeline, should be semantically versioned (e.g., v1.2.2 to v1.8.1).
Laboratories also should document the versions of the individual components of the pipeline. If a laboratory develops and manages one or more pipeline components, it should follow the same version control principles as the entire pipeline. Since pipeline upgrades often significantly change the NGS test results (e.g. ability to detect new variant types, change in report format) and the clinical report content, it is a good practice to communicate such changes to clinical teams and clients.
Reporting NGS Test Results
Variant Nomenclature
Variant nomenclature is an essential part of a clinical report and represents the fundamental element of a molecular test result. The Human Genome Variation Society (HGVS) variant nomenclature system is the de-facto representation of sequence variants in a clinical report, which is universally accepted as a standard by laboratory accreditation agencies and understood by molecular professionals, clinicians, and medical genetics professionals (13). The synthesis of this nomenclature for variants identified by NGS testing requires a complex process of conversion of the coordinate system from the reference genome to specific complementary DNA and protein transcripts. The alignment of the transcripts to the forward or the reverse genomic DNA strands and the HGVS 3’rule for variants in repeat sequence regions add additional complexity to the process. Several annotation tools—both open source and commercial—can generate HGVS nomenclature. However, since they might render inconsistent HGVS nomenclature, laboratories need to optimize and validate them for clinical use.
Variant Identification and Manipulation
The ability to detect sequence variants determines the performance characteristics for both a bioinformatics pipeline and an overall NGS assay. Several aspects of the pipeline can impact performance characteristics and affect the sensitivity of variant detection.
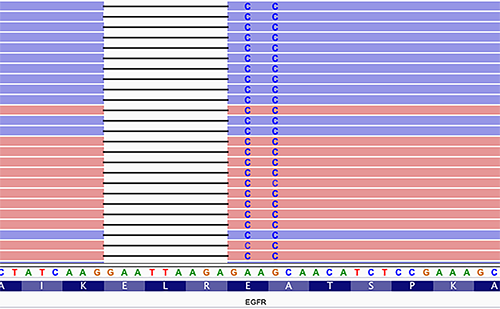
Estimating a pipeline’s false-negative rate accurately can be challenging. Identifying phased variants is one of the challenges. For example, EGFR inframe mutations in exon 19, which render tumors sensitive to tyrosine kinase inhibitors, are often identified as multiple variants that can be a variable combination of single nucleotide variants and insertions and deletions (Figure 3). Such horizontally complex variants represent a haplotype, in which the individual variants (primitives) are in-phase, i.e., present on the same contiguous sequencing reads (1). The correct identification of complex variants is vital for accurate clinical reporting and follow-up molecular testing on tumor relapse, including minimal residual disease testing. A limited number of variant calling algorithms are haplotype-aware, so laboratories should carefully review their variant calling algorithms during validation. VarGrouper is a relatively recent software tool that was developed to primarily address the limitation of variant calling algorithms without haplotype-aware variant detection features (14).
Click to View Full Size Image
Another contributor to the pool of false-negative (missed) variants is the process of in-silico masking of targeted NGS panels. Clinical laboratories commonly mask a portion of an NGS panel to report a subset of genes on the panel. This cost-effective approach is optimal for assay maintenance. Masking involves using tools that intersect a variant call file with a browser extensible data (BED) file that defines the regions of interest of the smaller reportable panel. The typical behavior of these algorithms results in an empty but perfectly valid variant call format (VCF) file in the absence of an input BED file. Consequently, for clinical testing, accidentally missing a BED file due to inconsistent transfer from development to the production environment may produce false-negative results and significantly impact patient care. Laboratories should design their pipelines to handle such intrinsic algorithm behavior and alert end-users to unexpected results in clinical testing.
Making a Scalable and Efficient Pipeline
Software Containers
The multiple components of a bioinformatics pipeline frequently have dependencies on different software run-times and in some instances, different versions of the same software. This results in a complex software ecosystem with unnecessary maintenance overhead, lack of portability, inconsistencies between development and production environments, and increased chance of errors. The integration of the pipeline with other software systems can also be challenging.
Software container technology has revolutionized the practice of software development and deployment across the globe. Containers are a standard unit of software that enables the packaging of software and its dependencies to be run on different computers and operating systems with virtually no configuration changes. Unlike virtual machines, containers are a lightweight Linux operating system process that isolates the software running inside the container from all other running applications on the computer. This allows portability across different IT platforms in healthcare systems and the cloud and avoids software conflicts.
Since containers are typically small software units, they can be instantiated very quickly to execute a specific task. A recent study demonstrated the distinct advantage of using containers for the bioinformatics pipeline such that NGS data analyzed on various IT infrastructures and with different workflow managers produced the same results (15). Containers also help implement version control. Containers are available as different open-source projects such as Docker, Singularity, Rkt, and LXC. Docker container is the most widely used of the general-purpose application containers. Singularity containers are designed specifically for bioinformatics applications on high-performance computing cluster systems.
Containers also provide a framework in which each step of the pipeline is provisioned into a container or application service. This enables an individual component of the pipeline to be updated in isolation without impacting other components.
Similarly, different pipeline components can be horizontally scaled to remove performance bottlenecks. A sophisticated software application that is deployed using several containers is typically managed in a production environment using container orchestration platforms such as Kubernetes, Mesos, Docker Swarm, and cloud vendor-specific frameworks.
Automation
Automation helps manage bioinformatics resources and workflows and streamlines day-to-day bioinformatics operations. The life-cycle of pipeline development, from testing and deployment to production infrastructure, is a complicated task. A laboratory must revalidate any upgrades to its pipeline to prevent unintended effects on test results. This manual testing and validation is time-consuming and, in some instances, inconsistent. The advantages of automation include more thorough and consistent enforcement of validation policies, regular testing and validation of pipeline upgrades, standardized version control, codebase integration, and proper documentation of audit trails for regulatory compliance.
An important use case of automation is the real-time monitoring of deployed bioinformatics pipelines in production. Edge-case scenarios related to the nature of sequencing data or unexpected changes in the deployment environment can significantly, often silently, impact NGS test results. In such scenarios, continuous monitoring and automated alert mechanisms are critical to avert unanticipated downtimes and erroneous test results. However, these advantages of automation come with a burden: time for initial setup and the learning curve of the bioinformatics team with automation tools.
Somak Roy, MD, is an assistant professor and director of genetic services, molecular informatics and director of the molecular genetic pathology fellowship program in the division of molecular and genomic pathology at the University of Pittsburgh Medical Center in Pittsburgh, Pennsylvania. +Email: [email protected]
References
- Roy S, Coldren C, Karunamurthy A, et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: A joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn 2018;20:4-27.
- Wang X, Liotta L. Clinical bioinformatics: A new emerging science. J Clin Bioinforma 2011;1:1.
- Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet 2019;10:426.
- Roy S, LaFramboise WA, Nikiforov YE, et al. Next-generation sequencing informatics: Challenges and strategies for implementation in a clinical environment. Arch Pathol Lab Med 2016;140:958-75.
- Hsi-Yang Fritz M, Leinonen R, Cochrane G, et al. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res 2011;21:734-40.
- Kadri S. Advances in next-generation sequencing bioinformatics for clinical diagnostics: Taking precision oncology to the next level. Advances in Molecular Pathology 2018;1:149-66.
- Abel HJ, Duncavage EJ. Detection of structural DNA variation from next generation sequencing data: A review of informatic approaches. Cancer Genet 2013;206:432-40.
- Kirchner M, Neumann O, Volckmar AL, et al. RNA-based detection of gene fusions in formalin-fixed and paraffin-embedded solid cancer samples. Cancers (Basel) 2019;11.
- Lubin IM, Aziz N, Babb LJ, et al. Principles and recommendations for standardizing the use of the next-generation sequencing variant file in clinical settings. J Mol Diagn 2017;19:417-26.
- Leipzig J. A review of bioinformatic pipeline frameworks. Brief Bioinform 2017;18:530-6.
- Fjukstad B, Bongo LA. A review of scalable bioinformatics pipelines. Data Science and Engineering 2017;2:245-51.
- Jennings LJ, Arcila ME, Corless C, et al. Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn 2017;19:341-65.
- Callenberg KM, Santana-Santos L, Chen L, et al. Clinical implementation and validation of automated human genome variation society (HGVS) nomenclature system for next-generation sequencing-based assays for cancer. J Mol Diagn 2018;20:628-34.
- Schmidt RJ, Macleay A, Le LP. VarGrouper: A bioinformatic tool for local haplotyping of deletion-insertion variants from next-generation sequencing data after variant calling. J Mol Diagn 2019;21:384-9.
- Kadri S, Roy S. Platform-agnostic deployment of bioinformatics pipelines for clinical NGS assays using containers, infrastructure orchestration, and workflow manager (Abstract #I031). J Mol Diagn 2019;21:1119–249.