It's a classic conundrum: biomarkers are essential to diagnosing, staging, treating, and monitoring cancer, yet despite an explosion of research, only a trickle have made it into clinical practice. The factors behind this less-than-desirable circumstance are complex at best, but according to some observers, boil down to the healthcare system's placing more value on cancer therapeutics relative to biomarkers. Without a better means of demonstrating the difference biomarkers make in clinical outcomes or management, they remain stuck in a loop of low value: inadequate funding for research that, in turn, limits the evidence for clinical utility, keeping their value low. A panel of leading scholars, clinicians, and executives recently collaborated about this dilemma in the hopes of starting a national dialogue toward breaking what they call a vicious cycle. Their proposed solutions—as ambitious as the problem is convoluted—can be implemented if the industry has the will to do so, they contend.
"People don't value tumor biomarkers. They value therapeutics. We all talk about personalized medicine but we don't really mean it if we're not willing to value biomarkers the way we value therapeutics," said Daniel Hayes, MD, Stuart B. Padnos professor of breast cancer research and clinical director of the breast oncology program at the University of Michigan Comprehensive Cancer Center in Ann Arbor. "However, if we insist on doing biomarker research the same way we do therapeutic research, it gets very expensive. But without putting the kind of money and research into it that's necessary to determine clinical utility, payers don't want to pay as much for a diagnostic as a therapeutic, and we're stuck in this vicious cycle."
Hayes has been on the forefront of thinking about how to bring more clinically meaningful biomarkers into cancer care, and he was the lead author of the group's commentary, which was published in the July 31, 2013 issue of Science Translational Medicine (Sci Transl Med 2013;196:1–7).
A Bad Track Record
Whether or not they concur with all elements of Hayes' and his colleagues' vicious cycle concept, many in the cancer field agree that biomarkers have an anemic batting average. The scientific literature is replete with reports of promising cancer biomarkers, but fewer than two dozen protein tumor markers have been cleared by the Food and Drug Administration (FDA)—only about half in the past decade—and not all have been embraced by clinicians. The same is true of lab-developed tests (LDTs).
While some paradigm-shifting cancer biomarkers with obvious clinical utility have been implemented rapidly in practice, others—be they FDA-cleared or LDTs—never really have found a niche. An example of the latter is UGT1A1 testing prior to starting irinotecan hydrochloride chemotherapy in colorectal cancer patients. Individuals who are homozygous for the UGT1A1*28 allele are at increased risk for neutropenia when taking irinotecan, but this testing just hasn't caught on, according to Hayes' co-author, Richard L. Schilsky, MD, chief medical officer of the American Society of Clinical Oncology (ASCO). "Practically no oncologist orders the test because it's not clear what you're supposed to do if you get a result back that shows your patient is high-risk. There's not a specific recommendation about whether you're supposed to omit the drug, lower the dose, lower the dose by how much, and whether lowering the dose actually mitigates the side-effects and whether or not it actually reduces the effectiveness of the treatment," he explained.
On the other end of the spectrum is KRAS genetic testing in metastatic colon cancer. After seminal research published in 2008 showed that patients without this mutation were more likely to respond to anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibody therapy, the test, even as an LDT, quickly became the de facto standard-of-care.
"A wealth of data came out all at the same time, and almost overnight oncologists started ordering KRAS testing and stopped prescribing the relevant drugs for patients whose tumors had KRAS mutations," recalled Schilsky. "That was a clear example of where if the tumor has a mutation, the drug doesn't work, and you shouldn't give it. That's the kind of discrete information that oncologists are always looking for."
The KRAS test also hit the medical economics jackpot: one anti-EGFR agent, cetuximab, costs anywhere from $110,000–160,000 per year, making an easy argument for reserving this treatment for patients most likely to benefit from it. At the same time, the cost of KRAS testing, about $400, speaks to Hayes' and his colleagues' arguments about the vicious cycle. If the test has that much clinical impact, shouldn't it be valued higher in the medical system?
Is Drug Development a Model?
"The pharmaceutical industry potentially could be a model for how to do biomarker validation and evidence generation. But then we'd have to charge a whole lot more for the tests, which everybody sees as commodities right now," said Hayes' co-author Debra Leonard, MD, PhD, professor and chair of pathology at the University of Vermont College of Medicine in Burlington. "Payers pay the cost of doing the test, but they aren't calculating in the cost of all that evidence generation. That's why drug companies can charge so much, because all the cost of generating evidence is built into the price of a drug when it comes onto the market. There's also good evidence to say that it does or doesn't work, and that's not the case for tests."
To Leonard's point about evidence, the authors cited one cause of the vicious cycle as how FDA regulates approvals for diagnostic tests (See Figure, below). The agency by statute does not have authority to require that proposed tests show clinical utility by improving clinical outcomes, but that is exactly what the authors would like to see. "In the current regulatory environment, many tumor-biomarker tests enter the market with analytical and clinical validity but insufficient information to establish their impact on healthcare outcomes. Thus, few of these tests are included in evidence-based guidelines, leaving healthcare professionals or third-party payers unsure of whether and how to use the tests or how much to pay for them," they wrote.
Vicious to Virtuous
A. Vicious Cycle
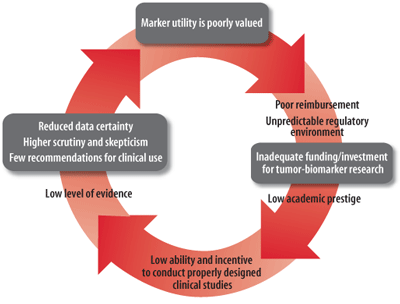
 (A) The vicious cycle of tumor-biomarker research and clinical utility.
(A) The vicious cycle of tumor-biomarker research and clinical utility.
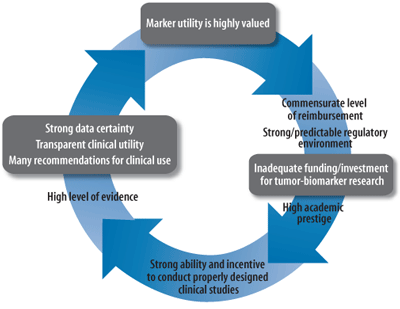
(B) A proposed virtuous cycle of tumor-biomarker research and clinical utility based on proposals herein.
Used with permission of Science Translational Medicine
Shaking Up the FDA
The authors proposed several solutions to this challenge, some more audacious than others. On the bold side, they suggested that FDA reorganize how it reviews oncology products, consolidating now separate drug and diagnostic reviews into a single oncologic product line managed jointly by the respective drug and biomarker divisions; in the case of the former, the Office of Hematology and Oncology Products in the Center for Drug Evaluation and Research, and in the latter, the Office of In Vitro Diagnostics in the Center for Devices and Radiological Health.
In the same vein, the authors proposed that FDA approve or clear tests only with rigorous evidence of both clinical utility and analytical validity, using ASCO level 1 evidence criteria.
If those recommendations might be long-term goals, possibly even requiring Congressional approval, others seem more approachable, but perhaps no less controversial. One is that FDA begins regulating LDTs. This contentious topic, under review for more than 3 years at FDA, would, the authors suggest, subject all proposed tests to a risk-based review process, regardless of the manufacturer or commercialization strategy behind them. However, not even all the authors back the idea.
"I don't believe we can do away with LDTs," said Leonard. "If the FDA had a better mechanism for looking at LDTs in their risk-based system, that might be helpful. But I worry about everything having to go through FDA because of the slowness of the FDA process and the expense of using the FDA process. There also doesn't seem to be any idea of how we're going to get from where we are today to an FDA approval process that actually works."
Leonard also expressed skepticism that the FDA approval process inherently produces better or more clinically useful tests than do LDTs. "The FDA process does not look at clinical utility, and there is no evidence that FDA-cleared or -approved tests do any better when they get into clinical practice than ones that haven't gone through the FDA process and are LDTs," she said, citing the Health and Human Services Secretary's Advisory Committee on Genetics, Health, and Society, which, in its 2008 report on the U.S. system of overseeing genetic testing found a "paucity" of information about the clinical utility of genetic testing.
Other researchers who have thought about how to speed up the biomarker pipeline also find this recommendation troubling. "Basically eliminating LDTs, especially if this were not accompanied by a prior increase in reimbursement and research dollars, would be extremely negative," said Leigh Anderson, PhD, CEO of Washington, D.C.-based SISCAPA Assay Technologies. "I'm involved in collaborative work with a number of groups trying to develop new tests mainly in the cancer area and I don't think any of them would be in the position to think seriously about going forward with those if the LDT route didn't exist. They would have to raise hundreds of millions of additional dollars to take that approach. It's not trivial to develop an LDT, but to say a proposed assay has to be treated as an FDA-cleared in vitro diagnostic [IVD] represents a significant additional barrier."
The Benefits of Biospecimen Banks
Although Anderson wasn't on board with the authors' proposal about LDTs, he lauded their recommendation that all drug registration trials maintain a biospecimen bank, funded by the sponsoring drug company, so that subsequent researchers could access the samples for prospective-retrospective studies. In fact, writing in a Clinical Chemistry opinion piece along with the journal's editor-in-chief Nader Rifai, PhD, he recommended that the National Institutes of Health develop a list of key clinical diagnostic questions prioritized by disease impact and linked to studies or medical centers with corresponding biospecimens. This "would allow a much more informed and productive application of the existing biomarker resources and would provide a much-needed basis for arguing for the enormous potential health-economic value of successful new tests," they wrote (Clin Chem 2013;59:194–7).
Along with the need to provide higher levels of evidence for candidate cancer biomarkers, the authors called for a significant ramp-up in biomarker research investment and higher reimbursement for tests that demonstrate clinical utility. In addition, they recommended that scientific journals adhere to higher standards in publishing tumor biomarker studies and be as willing to publish biomarker studies that have negative results as those with positive findings. Finally, they proposed that guideline bodies follow evidence-based recommendations for tumor biomarker test use.
Emulating the PET Registry
To up the ante on these sweeping reforms, the authors believe addressing them in concert is the only way to break the vicious cycle. But how can the healthcare industry essentially reinvent a new paradigm for better valuing cancer biomarkers when the elements of doing so are like a gyrating Medusa's head of knotty, seemingly intractable challenges? The authors agree the problem is too daunting if considered only in its entirety. But they and other experts suggest that several tangible actions could move the field along substantially without too much chaos or pain.
For example, in the area of building evidence that would open the door for better reimbursement for cancer biomarker tests, Schilsky envisions a tissue or blood test equivalent to the National Oncologic PET Registry (NOPR). This ground-breaking initiative managed by the American College of Radiology (ACR) and the ACR Imaging Network developed evidence for Medicare to reimburse PET scanning with F-18 fluorodeoxyglucose when it wasn't covered at all. NOPR enabled reimbursement for this testing in cancer patients on the proviso that physicians agreed to enter data in a registry that would enable a fair assessment of the impact PET had on cancer patient management. Started in 2006, NOPR led in 2009 to coverage of PET scans as part of the initial treatment strategy for most solid tumors, coverage that recently was expanded to include payment for up to three PET scans in patients with advanced cancer after their initial treatment, according to Schilsky.
By doing the same thing with selected tumor biomarkers, Schilsky suggested, "we immediately begin to capture information that we're not currently getting on the prevalence of use of certain tests, the kinds of clinical decisions based on those tests, and the outcomes of the patients who undergo the testing," he explained. "Then, for payers, it becomes much less of a Wild West environment. They will have information they can analyze and use to inform their coverage decisions." Such a system also would differentiate the most clinically useful tests from less relevant ones, enabling payers to shift resources to the winning tests. This, in turn, would incentivize test developers "to put tests out that are likely to perform well," Schilsky added.
The authors also point to efforts like the National Human Genome Research Institute's recent decision to fund more than $25 million over 4 years to develop the Clinical Genome Resource for authoritative information on genetic variants relevant to human disease and patient care. The National Cancer Institute (NCI) also is planning a web-based inventory of all biospecimens collected under its clinical trials cooperative group program, according to Schilsky.
The Impact of Technology
Anderson believes the authors overlooked the impact technological advances could have on the vicious cycle, by speeding up the process of vetting candidate biomarkers. "An alternative way of doing mass spectrometry [MS]-based protein assays is to analyze for specific targeted proteins in smaller numbers, so you might measure 10 or 100, but accurately and quickly," he explained. "Those kinds of directed assay methods which are not looking for everything, but instead for specific things that you hypothesize are important biomarkers, can be run fast enough and cheaply enough that you can run hundreds and hundreds of samples in a practical way. That then removes the primary technological limitation to getting the validation of biomarkers done."
Anderson added that this type of MS-based directed protein analysis also could speed up the bench-to-bedside time for biomarkers. "The advantage is that the same method used in biomarker verification studies at the research stage can be implemented at least in the large reference labs as LDTs, where it provides a significant technology improvement over immunoassays," said Anderson. "That it can be taken all the way from research to LDT in a capable reference lab takes a lot of delay out of the introduction into clinical practice. That's because you don't need to redevelop immunoassays for different platforms. Eventually you might not even need to redevelop it for an IVD platform, once mass spectrometry IVD platforms exist." He predicted that this approach could shave 5–10 years from the biomarker development process. Anderson's company, SISCAPA Assay Technologies, provides mass-spec-based specific assays for biomarker proteins.
Researchers also have a responsibility to think about the clinical need they want to address before diving deep into discovery, suggested Ivan Blasutig, PhD, a clinical biochemist and assistant professor at Toronto General Hospital and the University of Toronto. "Many of the biomarkers discovered may have statistically significant results but when it comes to actual clinical use they don't cut it. That's one of the biggest issues," he said.
Analytics: The Achilles Heel
Blasutig collaborates closely with Eleftherios Diamandis, MD, PhD, who has written extensively about the challenges of bringing proposed cancer biomarkers into clinical practice. He, Diamandis, and others have emphasized how important robust analytics are in the early stages of biomarker discovery. In fact Blasutig and Diamandis recently wrote about how using what turned out to be an unreliable commercial kit for CUZD1 detection set back their research team by 2 years and about $500,000 in their quest to find a new pancreatic cancer biomarker (Clin Chem 2013 doi:10.1373/clinchem.2013.215236).
Blasutig and others encouraged clinical laboratorians to participate actively in biomarker discovery, as they bring a wealth of knowledge about analytical issues in validation that research chemists don't necessarily share. Laboratorians also are more likely to be aware of resources like the NCI's Early Detection Research Network, which gives guidance on topics such as completing sample collections and avoiding analytical bias, Blasutig suggested.
If the vicious cycle seems completely unwieldy and unrepairable, each person CLN contacted expressed confidence that even in the face of long odds, it can be changed. Leonard spoke for many: "I'm optimistic that we have to do this for our patients, and for our healthcare delivery system. There are a lot of good people around who are interested in having this conversation. So if we can just get all the parties at the table and see if there are some concrete steps that we can take, that would be a major step in the right direction."