The current controversy about regulation of laboratory-developed tests (LDTs) is not new. The Food and Drug Administration (FDA) issued the Analyte Specific Reagent (ASR) Ruling in 1998 and the draft guidance for In Vitro Diagnostic Multivariate Index Assays (IVDMIA) in 2007, in similar attempts to regulate testing in clinical diagnostic laboratories. However, concerns about LDTs surfaced again several years ago, when in 2009, the College of American Pathologists (CAP) recommended that FDA play a role in the oversight of LDTs classified as high risk, arguing for stronger accreditation standards for laboratories conducting low- or moderate-risk LDTs. Although FDA held a public meeting in 2010 to hear stakeholder concerns, the agency has yet to issue new guidance.
This potential for FDA oversight of LDTs has sparked considerable concerns in laboratories that perform flow cytometry. Most current laboratory best practices and recommendations for validating tests were designed for traditional clinical chemistry assays and not for flow cytometry analyses. Furthermore, today's highly complex flow cytometers produce a wide variety of data outputs, and appropriate cellular reference materials are not available for validation of LDTs. Consequently, regulation of laboratory-developed flow cytometry tests could hamper the availability of tests using this technology.
In March 2011, international experts in the field of flow cytometry assay development and validation representing the International Council for Standardization of Haematology (ICSH) and the International Clinical Cytometry Society (ICCS) formed a workgroup to draft guidelines for validating flow cytometry LDTs. Their recommendations, "Validation of Cell Based Fluorescence Assays: Practice Guideline," were published recently (1).
This article will focus on the validation approach recommended in the ICSH/ICCS guidelines (1) for flow cytometric assays and highlight some of the differences with clinical chemistry method validations.
Laboratory Requirement for Validation
Validating an LDT method for any platform has many steps. The laboratory must establish the method performance specifications for accuracy, imprecision, analytical sensitivity, analytical specificity, reportable ranges, reference intervals, and any other performance characteristics required to test performance. The following discussion focuses on application of these principles to flow cytometry methods.
Categories of Bioanalytical Data
The first step of an analytical method validation should be to define the reportable results to be generated by the method and their respective data type. It is critical for a successful method validation to fully understand the category of bioanalytical data generated from the method and how the data type contributes to assay performance as well as assay limitations. In a 2005 Conference Report from the American Association of Pharmaceutical Scientists (AAPS), Jean Lee and others described four categories for bioanalytical data: quantitative, relative quantitative, quasi-quantitative, and qualitative (Table 1) (2).
|
Table 1
Categories of Bioanalytical Methods
|
| Assay Category |
Definition |
| Quantitative |
Uses calibration standard to determine the absolute quantitative values for unknown samples. The reference material is well defined and fully representative of the endogenous analyte.
Example: Insulin and steroid mass spec assays.
|
| Relative quantitative |
Uses a calibration standard to estimate the absolute quantitative values for unknown samples.
The reference material is not fully representative of the endogenous analyte.
Example: Flow cytometric methods in which antigen expression levels are quantified using ABC or MESF beads.
|
| Quasi-quantitative |
Does not use calibration standard, but has a continuous response. Numeric data is reported.
Example: Most flow cytometric methods reporting relative percentage and/or absolute counts of a specified cellular phenotype.
|
| Qualitative |
Lacks proportionality to the amount of analyte.
Categorical data is reported.
Example: Leukemia/lymphoma assays in which the phenotype of an abnormal cell type is described.
|
| Adapted from Lee et al. 2005 and O'Hara et al. 2011. |
Most flow cytometric methods fall into the quasi-quantitative category. In this case, results generated by the method are numeric and are proportional to the measured test sample characteristic, but they are not derived from a reference standard (Figure 1) (3). An example of this is the enumeration of CD4 T cells, one of the most common flow cytometric methods in clinical laboratories worldwide. In this multiplex assay, whole blood is directly stained with fluorescent conjugated antibodies to CD45 (for lymphocyte identification), CD3 (for T cell identification), CD4, and CD8 (to distinguish the two major T cell subsets). The events satisfying the definition of CD4 T lymphocytes (low side scatter, bright CD45 expression, CD3 positive, CD4 positive) are reported as the relative percentage of lymphocytes (low side scatter, bright CD45 expression). During the data analysis—or gating—each decision point in the process that identifies the CD4 T cells is based on properties of the test sample. These properties are numeric, such as the expression level of each antigen and the light scatter, but reference standards for antigen expression are not employed (Figure 2).
Figure 1
Enumeration of CD4+ T Cells From a Lymphocyte and Monocyte Gate
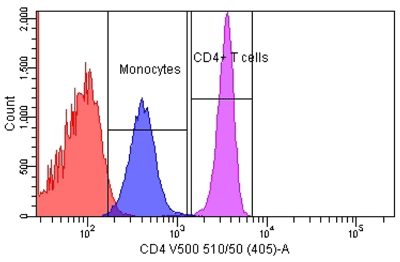 Peripheral whole blood was stained with fluorescent-conjugated mono-clonal antibody to CD4 in a lyse-wash procedure. Lymphocytes and monocytes were enumerated to 80,000 events using a BD FACSCantoII equipped with 405, 488, and 633 lasers. The sample was analyzed using FACSDiva Version 6.1.2 software to illustrate the relative number of bright CD4+ T cells and dim CD4+ monocytes. Reference standards were not employed.
Peripheral whole blood was stained with fluorescent-conjugated mono-clonal antibody to CD4 in a lyse-wash procedure. Lymphocytes and monocytes were enumerated to 80,000 events using a BD FACSCantoII equipped with 405, 488, and 633 lasers. The sample was analyzed using FACSDiva Version 6.1.2 software to illustrate the relative number of bright CD4+ T cells and dim CD4+ monocytes. Reference standards were not employed.
Flow cytometric methods that quantify antigen expression levels using quantitative fluorescence calibration beads are considered relative quantitative. These methods estimate antibodies bound per cell or molecules of equivalent fluorescence. The diagnostic test for sepsis and inflammation based on the quantitation of CD64 expression levels on peripheral blood neutrophils is an example of a relative quantitative, flow cytometric method (4).
Other flow cytometric methods in the clinical laboratory, such as leukemia lymphoma panels, are generally qualitative methods. These methods identify and characterize aberrant cellular phenotypes, and the data are interpreted by a qualified hematopathologist in order to diagnosis a particular pathology.
Figure 2
Identification of Different T Cell Subpopulations
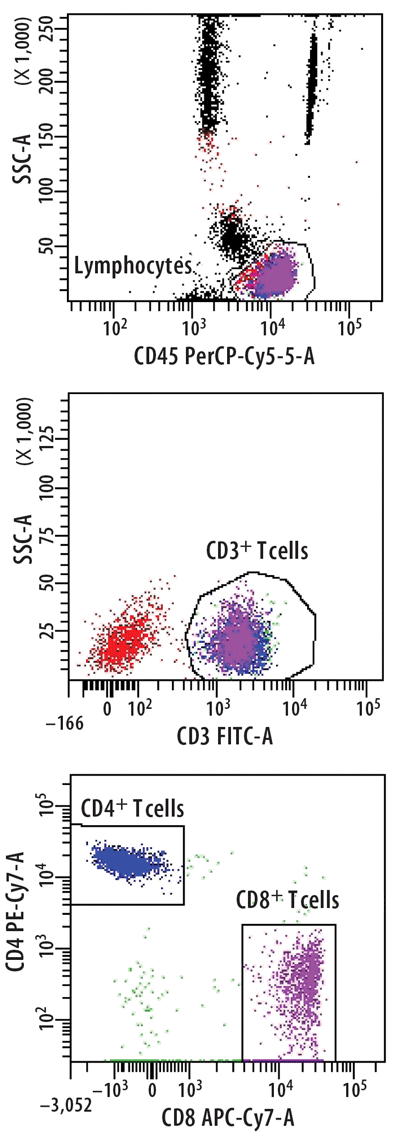 T cells were stained from peripheral whole blood with indicated fluorescent-conjugated monoclonal anti-bodies in a lyse-no wash procedure. Lymphocytes were enumerated to 80,000 events using a BD FACSCantoII equipped with 405, 488, and 633 lasers. The sample was analyzed using FACSDiva Version 6.1.2 software to illustrate the relative percentages of CD4+ T cells or CD8+ T cells based on the expression level of each antibody and light scatter. Reference standards were not employed.
T cells were stained from peripheral whole blood with indicated fluorescent-conjugated monoclonal anti-bodies in a lyse-no wash procedure. Lymphocytes were enumerated to 80,000 events using a BD FACSCantoII equipped with 405, 488, and 633 lasers. The sample was analyzed using FACSDiva Version 6.1.2 software to illustrate the relative percentages of CD4+ T cells or CD8+ T cells based on the expression level of each antibody and light scatter. Reference standards were not employed.
Challenges Specific to the Validation of Flow Cytometric Methods
While flow cytometry offers enormous flexibility and the potential to identify a wide variety of cellular subsets, the lack of reference material with target values for each of these novel applications makes method validation challenging. As opposed to soluble analytes, the measurands in flow cytometry are primarily cellular targets defined by complex immunophenotyping and light scatter properties. As a result, validation parameters are very different from most automated/clinical chemistry methods, especially specificity, accuracy, sensitivity, linearity, and the use of assay controls.
Specificity
In a general clinical chemistry method validation, laboratories have a variety of different strategies to choose from to establish method specificity (5). Specificity may be budgeted as a portion of the systematic error—as determined by physiological considerations or clinical expertise—or tests may be evaluated independently for interference caused by icterus, hemolysis, and lipemia. Laboratories may also opt to accept the interference claims as published in the manufacturer's package insert.
In contrast, laboratories establish the specificity for flow cytometric LDTs during the assay development phase. Phenotypic definition of the population of interest, monoclonal antibody clone selection, fluorochrome assignment, panel design, and gating strategy all influence assay specificity. In addition, instrument setup and compensation have a tremendous impact on assay specificity (6).
If the assay objective is to identify a particular cellular subset in peripheral whole blood, the phenotype of that cellular subset must be well established in the literature. For example, memory B cells are defined as CD19+ lymphocytes, which co-express CD27 (7). Theoretically, a flow cytometric method for the identification of memory B cells would only require two fluorescent-conjugated monoclonal antibodies (mAb) specific for CD19 and CD27; however, that assay would not be highly specific, as CD19 cells represent as low as 2.5% of the lymphocytes in healthy individuals and T cells also express CD27. The inclusion of a fluorescent-conjugated mAb specific for CD45 in the panel will allow for better identification of lymphocytes from other leucocytes than use of light scatter properties alone. The inclusion of fluorescent-conjugated mAb specific for non-B cell antigens such as CD3, CD14, and CD56 will prevent the contamination of the B cell gate with other cell types (Figure 3). The correct pairing of antigens and fluorochromes as well as a proper instrument setup and compensation will prevent compensation artifacts and other spurious results.
Figure 3
Memory B Cell Identification: 2-color vs. 4-color Fluorescence
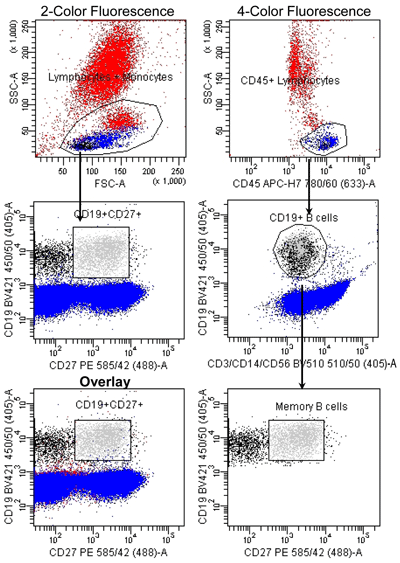 Peripheral whole blood was stained with indicated fluorescent-conjugated monoclonal antibodies in a lyse-wash protocol. B cells were enumerated (100,000 events) using a BD FACSCantoII equipped with 405, 488, and 633 lasers. Data was analyzed using FACSDiva Version 6.1.2 software. Analysis of the memory B cells directly from the lymphocyte gate determined 2,607 events were memory B cells (bright green). If analysis of memory B cells includes not only the CD45+ lymphocytes, but also discarding the cellular subsets of CD3+ T cells, CD14+ monocytes, and CD56+ NK cells, it allows better identification of our population resulting in a reduction of approximately 14% of non-memory B cells. If we then overlay these two graphs we get a visual picture of how many more non-memory B cells were included in the first analysis. Reference standards were not employed.
Peripheral whole blood was stained with indicated fluorescent-conjugated monoclonal antibodies in a lyse-wash protocol. B cells were enumerated (100,000 events) using a BD FACSCantoII equipped with 405, 488, and 633 lasers. Data was analyzed using FACSDiva Version 6.1.2 software. Analysis of the memory B cells directly from the lymphocyte gate determined 2,607 events were memory B cells (bright green). If analysis of memory B cells includes not only the CD45+ lymphocytes, but also discarding the cellular subsets of CD3+ T cells, CD14+ monocytes, and CD56+ NK cells, it allows better identification of our population resulting in a reduction of approximately 14% of non-memory B cells. If we then overlay these two graphs we get a visual picture of how many more non-memory B cells were included in the first analysis. Reference standards were not employed.
Accuracy
A common definition of accuracy is how closely an average value obtained from a large series of test results agrees with an accepted reference value. With the advances in assay standardization processes and the availability of certified reference materials for soluble analytes, laboratories can easily establish accuracy for many assays for soluble analytes. Additionally, proficiency surveys are available for many laboratory tests. These surveys allow laboratories to perform method comparisons between instruments and other laboratories and evaluate the test method with a reference method when available. For research-use-only tests that measure soluble analytes, accuracy by spike and recovery or split testing with the manufacturer are appropriate options for accuracy assessment.
For flow cytometric methods, accuracy assessment for LDTs presents a major challenge. Although control materials consisting of preserved whole blood are commercially available, they are generally calibrated to CD4 T cells and provide target values for only the major lymphocyte subset: CD3 T lymphocytes, and CD4 T, CD8 T, CD19, and CD16/CD56 NK cells. Moreover, the ranges manufacturers provide for target values are so large that they are not useful for assessing accuracy, and target values are not provided for complex subsets.
As previously mentioned, the remarkable strength of flow cytometry is the flexibility to detect a vast number of cellular subsets and intracellular antigens, even with the eight to 10 color clinical instruments. But this same flexibility makes it nearly impossible to develop cellular reference materials. The generally accepted alternative approaches for accuracy assessment such as the comparison to reference methodology, inter-laboratory comparison, and proficiency testing results are not applicable for novel flow cytometric methods. The ICSH/ICCS guidelines offer laboratories strategies for assessing accuracy for novel flow cytometric methods, such as verification with specimens obtained from patients with a confirmed diagnosis (8).
Imprecision
In contrast to accuracy, assessing assay imprecision is relatively painless for flow cytometry LDTs. A typical flow cytometric assay, or panel, identifies numerous cell types. For example, in addition to the memory B cells discussed above, total B cells, naive B cells, and plasma cells could also be reported from the same panel. The relative percentage and/or the absolute counts of each B cell subset might be reported as well. Although assay imprecision is achievable with coefficients of variation under 10% for most reportable results, the imprecision of less abundant cell types such as plasma cells will be higher. The laboratory then needs to consider the intended use of the data and whether or not higher imprecision affects that intended use. Another challenge in precision evaluation is finding validation samples with varying levels of each of the numerous potential reportable results.
Sensitivity
Common general clinical laboratory practices for sensitivity assessment include adopting the limit of the blank (LOB) and limit of detection (LOD) from the manufacturer's package insert. Strategies for verifying or establishing the lower limit of quantitation (LLOQ) at which the analyte can be reliably measured vary by laboratory and are detailed elsewhere (9). Laboratories can establish detection limits by verifying the package insert, by medical decision cut points, or as specified by clients or the laboratory director.
Although not typically part of a flow cytometric method validation, establishing a LOB/LOD and LLOQ is useful for flow cytometry LDTs. Eliminating one or two mAbs from a flow cytometry panel can establish the LOB/LOD. In the example discussed above for the enumeration of CD4 T cells, the panel included anti-CD45-PerCP-Cy7, anti-CD3-FITC, anti-CD4-PE-Cy7, and anti-CD8-APC-H7. The laboratory could stain whole blood with all reagents except the anti-CD4-PE-Cy7 and create a "blank" for CD4 T cells. Then any events found in the CD4 T cell gate would be considered background events. The LOD could then be established as the mean + 3 standard deviations of the number of samples (9). An alternate approach for some assay types (non-counting methods) would be to look at internal cell populations with known absence of antigen expression, provided that large differences in auto-fluorescence levels of different populations are not evident.
For the LLOQ evaluation, a laboratory can create samples with very low levels of detectable CD4 T cells using a similar strategy: spiking varying amounts of whole blood from a fully stained sample into samples stained without anti-CD4-PE-Cy7, and making sure that unbound mAb has been removed by several wash steps. Another option is using 100-fold excess of "cold" antibody. The LLOQ is then established, as with any method, as the lowest level where acceptable imprecision is achieved.
Linearity
Clinical laboratories must verify the linear range of an assay as specified by the package insert for quantitative methods. While laboratories can employ various strategies, it is recommended that they evaluate at least 5–7 concentrations which span the analytical measuring range (10). As discussed by Lee et al., it is not possible to assess linearity with quasi-quantitative methods (2). Nonetheless, in flow cytometry, instrument linearity can still be demonstrated. The ICSH/ICCS guidelines propose the use of beads to demonstrate instrument linearity using voltage settings applied to a particular assay. The beads can be analyzed suspended in buffer or when spiked into assay samples (6).
Assay Controls
As discussed above, a major challenge in validating flow cytometric methods is the lack of reference standards and qualified QC materials. While reference material and assay controls, in general, are readily available for most clinical chemistry assays, this is not the case for flow cytometry LDTs. The ICSH/ICCS Guideline proposes the use of internal cell populations for positive and negative controls, provided that there is a scientific justification for this approach, such as defined cell populations which either lack or constitutively express the antigen of interest (6). For certain assays, such as those of a quasi-quantitative or quantitative nature, internal assay QC may be preferable to commercially available control materials which have inherent limitations. For example, external QC samples will not pick up pre-analytic issues with the patient specimen, and in some cases they may include modified or surrogate cellular constituents.
Summary
In this article, we have highlighted the major points brought forth from the ICSH/ICCS Workgroup on Cell Based Fluorescence Assays. In developing guidelines for method validation, understanding the technology and the bioanalytical data type are some of the most important points laboratories must consider. As flow cytometry and related imaging technologies continue to evolve, the benefits they bring to the clinical laboratory will only increase.
REFEREENCES
- Davis BH, Wood B, Oldaker T, et al. Validation of cell-based fluorescence assays: Practice guidelines from the ICSH and ICCS – part I – rational and aims. Cytometry Part B Clin Cytom 2013;84:282–5.
- Lee JW, Weiner RS, Sailstad JM, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: A conference report. Pharm Res 2005;22:499.
- O'Hara D, Xu Y, Lianz E, et al. Recommendations for the validation of flow cytometric testing during drug development: Section II assays. J Immunol Methods 2011; 363:120–34.
- Davis BH, Olsen SH, Ahmad E, et al. Neutrophil CD64 is an improved indicator of infection or sepsis in emergency department patients. Arch Pathol Lab Med 2006; 130:654–61.
- Clinical and Laboratory Standards Institute (CLSI). Interference testing in clinical chemistry; Approved guideline—second edition. CLSI EP7-A2 2005.
- Tanqri S, Vall H, Kaplan Z, et al. Analytical issues for validation of cell-based fluorescence assays. Cytometry Part B Clin Cytom 2013;84:291–308.
- Perez-Andres M, Paiva B, Nieto WG, et al.; Primary Health Care Group of Salamanca for the Study of MBL. Human peripheral blood B-cell compartments: A crossroad in B-cell traffic. Cytometry Part B Clin Cytom 2010;78 Suppl. 1:S47–60.
- Wood B, Jevremovic D, Bene MC, et al. Validation of cell-based fluorescence assays: Practice guidelines from the ICSH and ICCS – part V – assay performance criteria. Cytometry Part B Clin Cytom 2013;84:315–23.
- CLSI. Protocols for determination of limits of detection and limits of quantitation; Approved guideline. CLSI EP17-A 2004.
- CLSI. Evaluation of the linearity of quantitative measurement procedures: A statistical approach; Approved guideline. CLSI EP06-A 2003.
Acknowledgments
The authors wish to thank Bruce Davis and Teri Oldaker for critical review of the manuscript and Sarah Livingston for assistance with manuscript preparation.

Virginia Litwin, PhD, is a principal scientist in the Hematology Department, Covance Central Laboratories Services, Indianapolis, and co-editor of the book, "Flow Cytometry in Drug Discovery and Development."
Email: [email protected]

Janelle Salkowitz-Bokal, PhD, MBA, is a staff scientist in the Hematology Department, Covance Central Laboratory Services, Indianapolis.
Email: [email protected]

Pamela Steele, PhD, is the senior scientist in the Global Immunology Department, Covance Central Laboratory Services, Indianapolis.
Email: [email protected]
Disclosure: The authors have nothing to disclose.