
Severe combined immunodeficiencies (SCID) are a group of genetically heterogeneous disorders with a common clinical phenotype of profound susceptibility to life-threatening infections. If left untreated, most babies born with the disease die before the age of 2 years. The immunological phenotype primarily involves T-cell impairment, both quantitative and functional, which affects immune response. However, other components of the immune response, such as B and NK cells, may also be affected, depending on the underlying genetic defect.
Although the number of genetic defects associated with a SCID phenotype varies depending on the reporting source, researchers believe approximately 12 to 15 distinct gene defects may be responsible for the more typical forms of SCID. Other genetic defects also may be associated with either T-cell lymphopenia of variable severity or combined immunodeficiencies.
The advent of newborn screening for SCID in several states has changed the outlook on this disease. Previously, the incidence of SCID was thought to be approximately 1 in 100,000 births; however, current estimates, based on newborn screening, suggest the incidence is as high as 1 in 58,000, although this varies with ethnicity of the population studied. A confounding factor is that SCID is typically asymptomatic at birth in the majority of affected babies, and the diagnosis can be missed until the onset of symptoms at a median age of 4–7 months. The first signs include recurrent, severe, and persistent viral, bacterial, and fungal infections coupled with a failure to thrive.
Laboratory professionals should be familiar with newborn screening for SCID and the types of tests used. This article provides background information on T-cell biology, the role of T-cells in immune function, and the genetic defects associated with SCID. It also describes newborn screening and treatment of affected babies.
Immunobiology Primer
The common characteristic of all typical forms of SCID is the presence of T-cell lymphopenia along with impaired T-cell function. Therefore, it is helpful to understand how the disease pathology affects T-cell development in humans.
T cells develop from hematopoietic precursors originating in the bone marrow in the so-called "committed to the T-cell pathway." The majority of development takes place in the thymus. One of the fundamental events during T-cell maturation in the thymus is formation of the T-cell receptor (TCR) by a process of somatic recombination of specific gene segments, a phenomenon referred to as VDJ recombination. During TCR generation, a DNA "by-product" is generated called the T-cell receptor excision circle (TREC) and serves as a helpful marker of naive T-cell development and production in the thymus, especially in the neonate. Since these excision circles are extra-chromosomal, they are diluted by cell division in peripheral circulation (blood); therefore, caution must be used when using TREC as a primary diagnostic marker for recent thymic emigration.
Quantitation of TRECs, which is performed using molecular techniques, is also affected by the longevity of naive T cells in peripheral circulation, as documented by the presence of TREC-positive T cells several years after thymectomy. Measuring TRECs in blood, however, serves as a useful tool for rapid and early identification of T-cell lymphopenia in the neonatal period. A mature T cell that has completed all the developmental stages in the thymus and is ready for peripheral export expresses TCR, which includes the associated co-receptors, CD3 and CD4 or CD8, depending on the type of the T cell. The TCR governs the antigenic specificity of the T cell and provides the fundamental basis for T-cell function in the immune response, in addition to co-stimulatory signals provided by non-TCR receptors on T cells and cellular interactions with other cells, including antigen-presenting cells (APCs).
Mature peripheral T cells also express cell-surface receptors that characterize their status as naive or antigen-inexperienced in terms of immune activation. These phenotypic markers include CD45RA, CD62L, and CCR7. Antigen-experienced T cells, however, are considered memory T cells, especially if they form part of the long-term T-cell memory pool and express the cellular marker CD45RO in addition to other receptors.
The Role of T Cells
T cells are fundamental to the adaptive immune response and form the backbone of cellular immunity. They participate in direct killing of pathogen-infected host cells in addition to activating other components of the immune response, such as macrophages, to facilitate killing of intracellular pathogens. T cells also help B cells, the other key component of the adaptive immune response, to produce antibodies, especially to protein antigens (T-dependent antigens).
Genetic Defects
The genetics of SCID are complex, encompassing defects in several genes that result in common clinical and immunological features, the most important being a significant decrease in or absence of T cells and impairment in cellular immune function. In addition, researchers have described other genetic defects that, although not associated with such a profound clinical phenotype, produce varying degrees of T-cell lymphopenia.
The majority of SCID genetic defects are inherited, monogenic disorders. With the exception of X-linked SCID, or the "boy-in-the-bubble" defect (caused by mutations in the IL2RG gene, which encodes the common gamma chain and is critical for the production of key cytokines involved in T and NK cell development and maturation), the majority of typical SCID are autosomal recessive disorders. While T-cell deficiencies are a universal feature of SCID, other immune defects are also observed in certain types of SCID, such as B and/or NK cell quantitative and functional abnormalities.
Quantitative defects in the three major lymphocyte subsets—T, B, and NK cells—facilitate confirmation of T-cell lymphopenia and rapid triage of possible genetic defects. This analysis, called TBNK flow, can be performed in a diagnostic immunology or pathology laboratory by flow cytometry. Table 1 describes molecular categorization of SCID based on four major categories of TBNK quantitation. Fully penetrant mutations that result in complete loss of function of the associated protein (amorphic phenotype) cause typical or classic forms of SCID. Other mutations in the same genes may be hypomorphic and result in partial loss of function of the protein, producing the "leaky" SCID phenotype. When leaky SCID occurs in the clinical context of erythrodermia (skin rash), tissue inflammation, hepatosplenomegaly, eosinophilia, increased levels of serum IgE, oligoclonal autologous T cells, and diarrhea, the phenotype is called Omenn syndrome.
Table 1
Flow Cytometric Lymphocyte Subset Quantitation* |
TBNK phenotype by flow
cytometric quantitation |
Examples of gene defect |
| T-B+NK– |
IL2RG, JAK3 |
| T-B–NK+ |
RAG1, RAG2, DCLRE1C |
| T-B–NK– |
ADA, AK2 |
| T-B+NK+ |
IL7RA, PTPRC, CD3Z, CD3E, CD3D |
| *This analysis is used to identify potential SCID genetic defects. |
Table 2 provides a list of definitions which can be used to classify patients with SCID and T-cell lymphopenia, and Table 3 summarizes genetic defects associated with SCID, and leaky SCID/Omenn syndrome. In addition to the above-mentioned phenotypic and immunological spectrum, atypical forms of Omenn syndrome have been reported with only some of the features of this clinical phenotype. Thus far, atypical Omenn syndrome has been reported related to genetic defects in two genes associated with typical and leaky forms of SCID, IL2RG and ADA, which cause X-linked SCID and adenosine deaminase deficiency, respectively.
|
Table 2
Definition of SCID Categories
|
| SCID or T-cell lymphopenia category |
Basic definition |
| Typical SCID |
<300 autologous CD3 T cells/µL blood; requires immediate medical intervention. |
| Leaky SCID or Omenn syndrome |
300–1500 autologous CD3 T cells/µL blood (T-cell lymphocytosis in some patients); hypomorphic mutations in genes associated with typical SCID; Omenn syndrome must fulfill the clinical characteristics associated with that entity; requires medical intervention as with typical SCID. |
| Variant SCID |
A form of leaky SCID where the molecular defect is unknown; may or may not require intervention, such as hematopoietic cell transplantation. |
Syndromes with
T-cell lymphopenia |
Includes chromosomal deletion or duplication syndromes or other genetic defects associated with T-cell lymphopenia of variable severity (CD3 T cells typically ≤1500 cells/µL; may or may not have T-cell functional impairment, e.g., DiGeorge syndrome, CHARGE syndrome, Down syndrome, Ataxia telangiectasia, DOCK8 deficiency). |
| Secondary T-cell lymphopenia |
Includes contexts of T-cell lymphopenia (CD3 T cells typically ≤1500 cells/µL) related to non-genetic causes but excludes premature infants (e.g., intestinal lymphangiectasia, anasarca, chylothorax, third-spacing, cardiac surgery +/- thymectomy). |
| T-cell lymphopenia of prematurity |
T-cell lymphopenia of variable severity depending on the extent of prematurity (typically ≤1500 cells/µL) and with no other recognizable clinical context to support the T-cell lymphopenia. |
| Source: Adapted from CLSI document NBS06-A. |
When considering the genetic etiology of SCID and T-cell lymphopenia, it is important to note that the same genetic mutation may result in variable clinical phenotypes in siblings, likely related to gene modulation, such as epigenetic alterations and/or environmental influence. Furthermore, mutations in the same gene in unrelated individuals may cause varying degrees of T-cell lymphopenia and dysfunction. A third possibility is that mutations in different genes may give rise to similar clinical and immunological phenotypes due to their common effects on T-cell development, maturation, and function.
A notable example is mutations in the ADA gene, which cause variable phenotypes, ranging from typical SCID to partial late-onset SCID, depending on how the mutation influences ADA enzymatic activity (Table 3). Finally, T-cell deficiency may be progressive with age and not readily apparent at birth or in the first months of life.
Table 3
Gene Defects Associated With SCID and T-cell Lymphopenia |
| Disease |
Gene defect |
| Typical SCID |
ADA, CD3D, CDZ, CD3E, IL2RG, DCLRE1C, IL7RA, JAK3, PTPRC, RAG1, RAG2, PRKDC, other genes |
| Reticular dysgenesis |
AK2 |
| Coronin-1A deficiency |
CORO1A |
| Complete DiGeorge syndrome |
22q1.2 deletion, other defects |
| Leaky SCID/Omenn syndrome |
Partial-loss-of-function mutations in ADA, PTPRC, IL2RG, DCLRE1C, IL7RA, JAK3, LIG4, RAG1, RAG2, other genes |
| Cartilage hair hypoplasia (CHH) |
RMRP |
| Folate metabolism and cobalamin deficiencies |
MTHFD1, MTR, SLC46A1 |
| Variant SCID |
Unknown molecular defects |
| Syndromes with T-cell impairment |
DiGeorge syndrome (22q11.2 del, TBX1, 10pdel), Cernunnos (XLF deficiency due to mutations in NHEJ1), CHARGE syndrome (CHD7), Jacobsen syndrome (11q del), RAC2 deficiency (RAC2), DOCK8 deficiency (DOCK8), Ataxia telangiectasia (ATM), VACTERL association (unknown genes, multiple organ defects), Barth syndrome (TAZ), TAR syndrome (RBM8A; 1q2.1 del), Down syndrome (Trisomy 21), EEC syndrome (TP63) |
| Genetic defects that may be variably associated with T-cell lymphopenia, but are not likely to be identified by newborn screening for SCID. |
Late-onset ADA deficiency, CD3G, CD8 deficiency, CARD11, IL2RG (p.R222C mutation), IKZF1 (IKAROS), ITK, LCK, MAGT1, bare lymphocyte syndrome I and II (TAP1, TAP2, TAPBP, CIITA, RXANK, RFX5, RFXAP), MST1, ORA1, STIM1, PNP, RHOH, STAT5B, TRAC, UNC119, ZAP70, CLOVES syndrome (PIK3CA) |
| Source: Adapted from CLSI document NBS06-A. |
Researchers continue to identify SCID-associated gene defects; however, the genetic etiology remains unknown for a subset of clinically classified SCID patients. Among the SCID-associated gene defects, mutations in the IL2RG, ADA, and IL7RA genes account for close to half of all typical cases, with X-linked SCID (IL2RG mutations) alone making up approximately 20% of all the typical SCID defects. It is important to note that specific ethnic populations have a higher incidence of certain types of SCID due to founder mutations. For example, ADA-SCID in the Somali population has an incidence of ~1 in 5,000, DCLRE1C (Artemis) gene mutations occur in Navajo Americans at a rate of ~1 in 2,000, and RAG1, RAG2, ADA, IL7RA, CD3, and ZAP70 mutations have a similar incidence in the Amish and Mennonite populations.
Newborn Screening
Given the significant impact of profound T-cell lymphopenia on the health and survival of affected infants, early diagnosis is essential to reducing morbidity and mortality. In fact, identification of the disease in newborns is considered a pediatric emergency.
Life-saving treatments are available, making SCID an attractive candidate for population-based newborn screening. In January 2010, the Secretary's Advisory Committee on Heritable Disorders in Newborns and Children voted to include SCID in the recommended uniform panel for newborn screening based on pilot data available from Wisconsin and Massachusetts, as well as the availability of a molecular assay to screen for thymic-derived T cells using PCR quantitation of TRECs in dried blood spots. As of September 2013, 16 states and the Navajo Nation have implemented state-wide newborn screening for SCID and T-cell lymphopenia. Three other states have been approved to start newborn screening shortly and additional states are likely to initiate pilot or routine newborn SCID screening in the next 12 months. As of May 2013, a little more than 3 million infants had been screened for SCID in the United States. The reported annual birth rate in the U.S. in 2012 was 63 births per 1,000 women. Therefore, SCID newborn screening has already captured more than half of the nation's annual births. The data from newborn screening in 10 states and the Navajo Nation is being collated and submitted for publication (Kwan A, et al. 2013, manuscript in preparation). The Newborn Screening Translational Research Network (NBSTRN) and the American College of Medical Genetics host several tools to facilitate data collection, quality control and assurance, analytical validation, and long-term follow-up for SCID newborn screening. For laboratorians, of particular interest is the Laboratory Data Collection and Improvement tool called the Region 4 Stork (R4S) SCID module supported by NBSTRN. This module can be accessed by public health laboratory professionals or other laboratory and clinical professionals who are actively involved in screening and want to contribute to the database. Collecting national and international newborn screening data in this module will help facilitate comparisons between screening laboratories and refinement of laboratory cutoffs for TREC values, as well as correlation of laboratory data with clinical information, including treatment, and/or genetic and immunological analyses.
In addition, the Clinical and Laboratory Standards Institute (CLSI) published a guideline in April 2013 on using TREC analysis in newborn screening, "Newborn Blood Spot Screening for Severe Combined Immunodeficiency by Measurement of T-cell Receptor Excision Circles, Approved Guideline NBS06-A." This document is a valuable reference for screening laboratories, diagnostic laboratory professionals, and clinicians.
Screening laboratories perform TREC analysis using a quantitative, real-time PCR assay (Figure 1). Several methods are used, which are discussed in detail in the CLSI document. The Centers for Disease Control and Prevention's Newborn Screening Branch also disseminates calibrators, quality control, and proficiency testing materials.
Figure 1
Newborn Screening for SCID
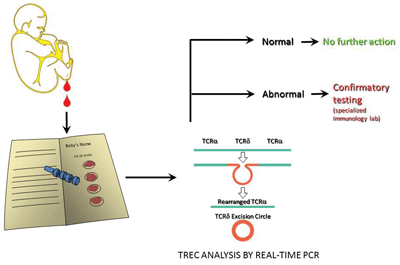 Newborn screening for SCID begins with analysis of a dried blood spot collected at birth. Babies with a positive result should have additional confirmatory tests. Current estimates suggest the incidence of this group of immunodeficiency disorders is as high as 1 in 40,000 births.
Newborn screening for SCID begins with analysis of a dried blood spot collected at birth. Babies with a positive result should have additional confirmatory tests. Current estimates suggest the incidence of this group of immunodeficiency disorders is as high as 1 in 40,000 births.
While TREC analysis has been very useful in identifying many types of SCID and other T-cell lymphopenias in newborns, researchers have reported that some forms of partial SCID are missed by this method, specifically ADA deficiency. However, mass spectrometric analysis of adenosine metabolites, including 2'-deoxyadenosine in dried blood spots, identified those cases. TREC-based newborn screening may also miss immune deficiencies other than SCID, such as Bare Lymphocyte Syndrome (due to MHC class I or II deficiency), where the T-cell lymphopenia is confined to specific T cell sub-populations, if the T-cell lymphopenia occurs later than the neonatal or early infancy period, or there is impaired T cell function in the context of normal T cell counts.
Follow-Up on SCID Babies
To provide immediate care for infants who have an abnormal TREC result, an efficient and rapid means of providing both short- and long-term follow-up must be in place. Effective communication between the state or other laboratory performing the newborn screening, pediatricians, and clinical immunologists with expertise in the care and treatment of these diseases is essential.
It is also important to keep in mind that an abnormal TREC result in newborn screening does not on its own constitute a diagnosis of SCID. Premature infants may have T-cell lymphopenia associated with prematurity that may correct itself when the baby reaches the appropriate gestational age (37 weeks) or may require additional evaluation for persistent T-cell lymphopenia. Also, newborn screening with the TREC assay has been shown to identify infants with other causes of T-cell lymphopenia apart from SCID such as DiGeorge syndrome, Down syndrome, CHARGE syndrome, and idiopathic T-cell lymphopenia. Furthermore, other non-genetic causes of T-cell lymphopenia, known as secondary T-cell lymphopenia (e.g., anasarca, intestinal lymphangiectasia, chylothorax, and cardiac surgery, among others), should be considered when pursuing further evaluation.
Laboratories perform confirmatory testing for SCID or other causes of T-cell lymphopenia using several methods: flow cytometric analysis of lymphocyte subsets, including T, B, and NK cells; quantitation of naive and memory T cells; T-cell functional studies such as in vitro proliferation assays for lymphocyte function; enzyme activity measurements of ADA levels; and genetic analysis to identify specific mutations. Depending on the clinical context, other diagnostic laboratory studies may also be needed, including, but not limited to, immunoglobulin quantitation, maternal engraftment assessment, evaluation for cytokine signaling defects, and radiation sensitivity analysis.
Given the complexity of these tests and the problems associated with obtaining blood from newborns, care plans should include access to immunology and molecular genetics laboratories that perform validated tests using only a small amount of blood. In addition, several pre-analytical considerations need to be kept in mind for such sensitive analyses and these are detailed in the CLSI guideline on SCID newborn screening.
Treating SCID
Treating infants with SCID or severe T-cell lymphopenia not only involves curative modalities but also practical management approaches that reduce the risk of mortality from infection. These strategies include: protective isolation to the extent possible; avoidance of live attenuated vaccines; breast-feeding only after the mother is confirmed to be seronegative for cytomegalovirus (CMV); use of leukodepleted, irradiated, CMV-negative blood products for transfusion; prophylactic antibiotics; and immunoglobulin replacement therapy, as needed.
For infants with a confirmed diagnosis, immune reconstitution via hematopoietic cell transplantation (HCT) is the most beneficial approach. There are several options for performing HCT, depending on the underlying clinical condition and availability of donors. In general, the earlier HCT is performed, the better the outcome, especially with regard to long-term survival. HCT is a complex treatment, and risks include acute or chronic graft-versus-host disease, chemotherapy toxicity, delayed or incomplete immune reconstitution, and autoimmunity. If severe, any of these can be fatal.
Other than HCT, enzyme replacement therapy with polyethylene glycol-conjugated ADA is an option for infants with ADA deficiency. This therapy may provide partial immune recovery until other treatment options become available or the patient becomes non-responsive to the enzyme therapy.
Another emerging treatment for SCID is gene therapy. Newer modalities have resulted in more effective immune recovery with less deleterious side effects, since earlier attempts at gene therapy resulted in development of leukemia in several patients as a side-effect of the treatment related to the vector used for the therapy. However, gene therapy is not available for all forms of SCID and only specific genetic defects, such as IL2RG and ADA, can potentially be treated with this approach.
The Outlook for SCID Screening
In summary, SCID and severe T-cell lymphopenia encompass a spectrum of immunological and clinical phenotypes that require urgent and effective intervention to reduce mortality. Population-based newborn screening followed by appropriate diagnostic testing offers a valuable and critical resource to both physicians and patients for accurate diagnosis and management. This is an area of rapid growth, discovery, and improvement in laboratory testing and also provides a paradigm for close and effective collaboration between diagnostic laboratory scientists and clinicians in improving patient care.
SUGGESTED READINGS
Brown L, Xu-Bayford J, Allwood Z, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: The case for newborn screening. Blood 2011;117:3243–6.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Ann Rev Immunol 2004;22:625–55.
Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol 2012;129:597–604.
Chan A, Scalchunes C, Boyle M, et al. Early vs. delayed diagnosis of severe combined immunodeficiency: A family perspective survey. Clin Immunol 2011;138:3–8.
Clinical and Laboratory Standards Institute (CLSI). Newborn blood spot screening for severe combined immunodeficiency by measurement of T-cell receptor excision circles; Approved guideline. CLSI NBS06-A 2013.
Kuo CY, Chase J, Lloret MG, et al. Newborn screening for severe combined immunodeficiency does not identify bare lymphocyte syndrome. J Allergy Clin Immunol 2013;131:1693–5.
la Marca G, Canessa C, Gioclaiere E, et al. Tandem mass spectrometry, but not T-cell receptor excision circle analysis, identifies newborns with late-onset adenosine deaminase deficiency. J Allergy Clin Immunol 2013;131:1604–10.
Lipstein EA, Vorono S, Browning MF, et al. Systematic evidence review of newborn screening and treatment of severe combined immunodeficiency. Pediatrics 2010;125:e1226–35.
Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles. J Allergy Clin Immunol 2012;129:607–16.
Shearer WT. Screening for severe combined immunodeficiency in newborns. J Allergy Clin Immunol 2012;129:619–21.

Roshini S. Abraham, PhD, is associate professor of Medicine and Laboratory Medicine and Pathology in the College of Medicine, Mayo Clinic, director of the Cellular and Molecular Immunology Laboratory within the Clinical Immunology Laboratories in the Department of Laboratory Medicine and Pathology, and director of the Jeffrey Modell Foundation for Primary Immunodeficiencies Diagnostic and Research Center at the Mayo Clinic in Rochester, Minn.
Email: [email protected]
Disclosure: The author has nothing to disclose.